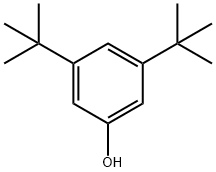
3,5-DI-TERT-BUTYLPHENOL synthesis
- Product Name:3,5-DI-TERT-BUTYLPHENOL
- CAS Number:1138-52-9
- Molecular formula:C14H22O
- Molecular Weight:206.32
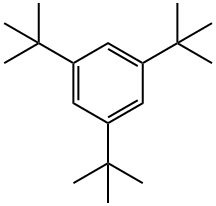
1460-02-2
219 suppliers
$6.00/5g
1138-52-9
152 suppliers
$8.00/100mg
Yield:1138-52-9 90%
Reaction Conditions:
Stage #1:1,3,5-Tri-tert-butylbenzene with iodine;magnesium in tetrahydrofuranInert atmosphere;Reflux;
Stage #2: with Trimethyl borate in tetrahydrofuran at 0 - 20;Inert atmosphere;
Stage #3: with Oxone in water;acetone at 20; for 1.11667 h;Catalytic behavior;Reagent/catalyst;Concentration;Temperature;Solvent;
Steps:
Synthesis of 3 via Borylation and Oxidation
A 250 mL round-bottom flask equipped with a Teflon-coatedmagnetic stir bar was charged with 8 (26.9 g, 100 mmol, 1equiv), Mg turnings (3.65 g, 150 mmol, 1.5 equiv), and a piece ofI2 crystal. The reaction was put under N2, and dry, degassed THF(100 mL) was added to the flask. The mixture was stirred at r.t.until initiation of the reaction was visible. An ice bath was usedfor cooling the reaction in the event that the temperature rosetoo high. In a separate 500 mL round-bottom flask equippedwith a Teflon-coated magnetic stir bar, B(OMe)3 (21.2 mL, 200mmol, 2 equiv) was added via syringe, followed by the additionof THF (100 mL). The solution was cooled to 0 °C using an icebath, and stirred under N2. The THF solution of the Grignardreagent was transferred to the cooled flask containing theB(OMe)3 solution via cannula. Once the addition was complete,the reaction was stirred vigorously and warmed back to r.t. for 2h. The reaction was then cooled back down to 0 °C andquenched by the addition of 1 M HCl (80 mL). The solution wasextracted using EtOAc (3 × 200 mL), the organic layers werecombined, and dried over MgSO4. The crude reaction mixturewas concentrated in vacuo to give a yellow solid. This was transferredto a 2 L round-bottom flask equipped with a Tefloncoatedmagnetic stir bar, re-dissolved in acetone (600 mL), andan aq solution of Oxone (61.5 g, 200 mmol, 2 equiv in 600 mLH2O) was added dropwise at r.t. over 1 h, and stirred for another7 min. The reaction was then carefully quenched by the additionof sat. aq NaHSO3 (200 mL), and concentrated in vacuo toremove most of the acetone. The crude reaction mixture wasextracted with CH2Cl2 (3 × 200 mL), the organic layers werecombined, dried over MgSO4, and concentrated in vacuo toobtain a yellow solid. Flash column chromatography (hexanes-EtOAc, gradient from 100:0 to 90:10) yielded 3 as a yellow solid(18.7 g, 90% before recrystallization), and it was recrystallizedfrom hexanes (50 mL) to yield pure 8 as colorless crystals (16.5g, 80%). 1H NMR (500 MHz, CDCl3): δ = 7.00 (t, J = 1.7 Hz, 1 H),6.68 (d, J = 1.7 Hz, 2 H), 4.52 (br s, 1 H), 1.30 (s, 18 H) ppm. 13CNMR (125 MHz, CDCl3): δ = 154.8, 152.7, 115.2, 110.0, 34.9, 31.5ppm. Analytical data matches that of the commercially availablematerial.
References:
Kwon, Ohhyeon;Esguerra, Kenneth Virgel N.;Glazerman, Michael;Petitjean, Laurène;Xu, Yalun;Ottenwaelder, Xavier;Lumb, Jean-Philip [Synlett,2017,vol. 28,# 13,p. 1548 - 1553] Location in patent:supporting information
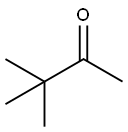
75-97-8
237 suppliers
$10.00/1g
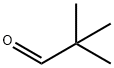
630-19-3
311 suppliers
$13.00/1g
1138-52-9
152 suppliers
$8.00/100mg
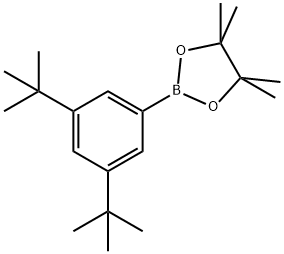
1071924-13-4
37 suppliers
inquiry
1138-52-9
152 suppliers
$8.00/100mg
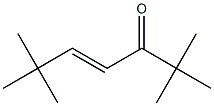
1653-94-7
0 suppliers
inquiry
1138-52-9
152 suppliers
$8.00/100mg
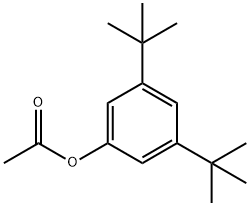
5723-95-5
1 suppliers
inquiry
1138-52-9
152 suppliers
$8.00/100mg