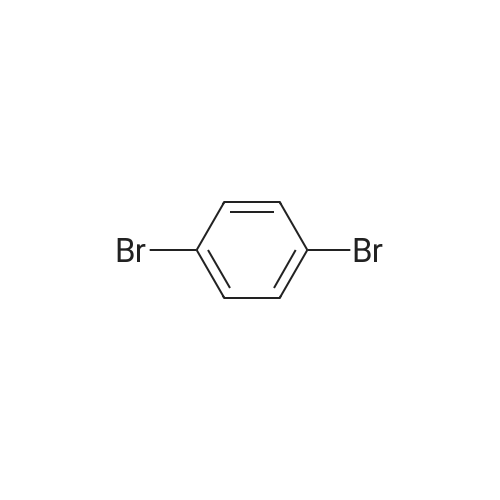
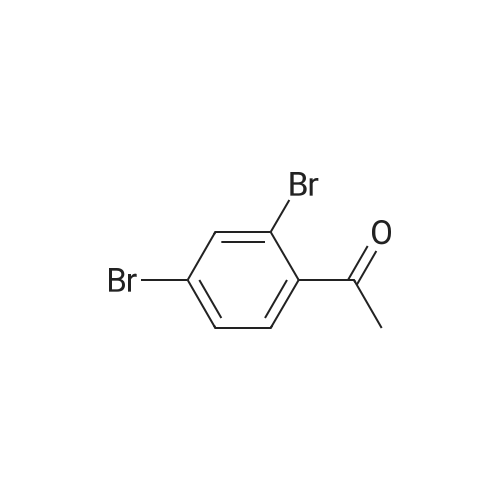
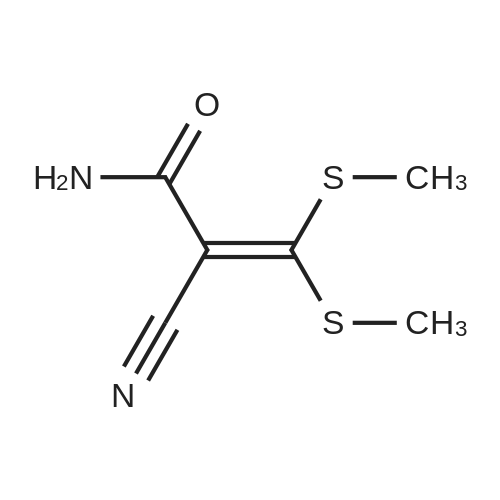
Abstract: Chiral cyclopropanols are highly desirable building blocks for medicinal chemistry, but the stereoselective synthesis of these molecules remains challenging. Here, a novel strategy is reported for the diastereo- and enantioselective synthesis of cyclopropanol derivatives via the biocatalytic asymmetric cyclopropanation of vinyl esters with ethyl diazoacetate (EDA). A dehaloperoxidase enzyme from Amphitrite ornata was repurposed to catalyze this challenging cyclopropanation reaction, and its activity and stereoselectivity were optimized via protein engineering. Using this system, a broad range of electron-deficient vinyl esters were efficiently converted to the desired cyclopropanation products with up to 99.5:0.5 diastereomeric and enantiomeric ratios. In addition, the engineered dehaloperoxidase-based biocatalyst is able to catalyze a variety of other abiological carbene transfer reactions, including N?H/S?H carbene insertion with EDA as well as cyclopropanation with diazoacetonitrile, thus adding to the multifunctionality of this enzyme and defining it as a valuable new scaffold for the development of novel carbene transferases.
Abstract: In this study, we developed a simple transition-metal-free borylation reaction of aryl bromides. Bis-boronic acid (BBA), was used, and the borylation reaction was performed using a simple procedure at a mild temperature. Under mild conditions, aryl bromides were converted to arylboronic acids directly without any deprotection steps and purified by conversion to trifluoroborate salts. The functional group tolerance was considerably high. The mechanism study suggested that this borylation reaction proceeds via a radical pathway.
* All experimental methods are cited from the reference, please refer to the original source for details. We do not guarantee the accuracy of the content in the reference.
With bis[chloro(1,2,3-trihapto-allylbenzene)palladium(II)]; C40H43BN2P(1-)*C16H32LiO4(1+); potassium <i>tert</i>-butylate In tolueneInert atmosphere; Schlenk technique
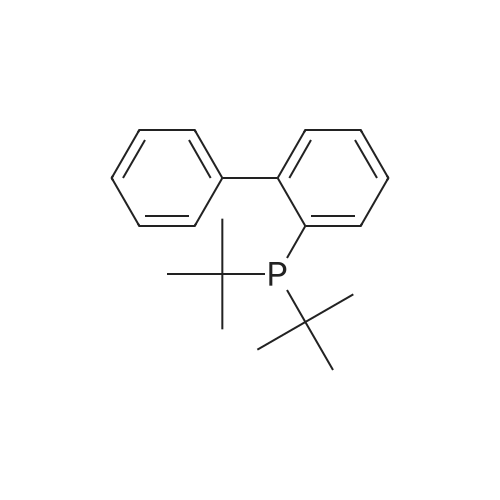
General procedure: Typically, [Pd(cinnamyl)Cl]2 (0.0031 g, 0.006 mmol), ligand 1 (0.0107 mg, 0.012 mmol), and KO(t-Bu) (0.081 g, 0.72 mmol) were loaded into a Schlenk tube. If a solid aryl bromide or amine was used, it was also added at this time. To the mixture of solids, the aryl bromide (0.6 mmol) and amine (0.72 mmol) were added via syringe (if liquid), followed by toluene (2 mL). The resulting mixture was stirred at room temperature for 1 min., then placed in a pre-heated, 80 °C oil bath and allowed to react for 12 h. After this time, the mixture was removed from the bath and cooled to room temperature, diluted with EtOAc (5 mL), and filtered through silica (1 × 4 cm column, ~10 mL), eluting with EtOAc (20 mL) or until the filtrate ran clear. The volatiles were removed from the filtrate via rotary evaporation and the resulting residue was subjected to flash chromatography on silica gel (8 × 2 cm column, ~25 mL silica). Specific details related to the synthesis, yield and characterization of each coupled product are described below in Section 4.8.
84%
With potassium phosphate; copper(l) iodide In diethylene glycol at 70℃; for 14 h; Sealed tube
General procedure: A 10 mL vial was charged with CuI (9.5 mg, 0.05 mmol), PSAP (30 mg,0.05 mmol, > 100 mesh), K3PO4 (424 mg, 2 mmol), aryl bromides (1mmol), amines (1.5 mmol), DEG (2 mL), and a magnetic stir bar. The vessel was sealed with a septum and placed into a preheated oil batchat 70 °C. The reaction mixture was held at this temperature for 14 hours. After cooling to r.t., the reaction mixture was filtered, and the precipitates were thoroughly washed with water and EtOAc (3 × 20mL). The combined organic phases were washed with water and brine, dried over anhydrous Na2SO4, and concentrated in vacuo. Theresidue was purified using flash column chromatography on silica gel(eluting with petroleum ether/EtOAc) to afford the desired products.
82%
With copper(l) iodide; tetrabutylammomium bromide; potassium hydroxide In water at 70℃; for 16 h; Green chemistry
General procedure: A 10 mL of vial was charged with CuI (10 mg, 0.05 mmol), PSP (0.25 mmol, size less than 90 μM), TBAB (40 mg, 0.25 mmol), base (1.0 mmol), aryl halides (0.5 mmol), arylamine (2.0 mmol), H2O (1.0 mL), and a magnetic stir bar. The vessel was sealed with a septum and placed into an oil bath, which was preheated to 70 °C (90 °C for alkyl amine, 120 °C for imidazole). The reaction mixture was stirred for another 16 h (8 h for imidazole). After allowing the mixture to cool to room temperature, the reaction mixture was filtrated, the precipitates were washed with water and ethyl acetate thoroughly. The filtrate was extracted with ethyl acetate (3×25 mL). The combined organic phases was washed with water and brine, dried over anhydrous Na2SO4, and concentrated in vacuo. The residue was purified by flash column chromatograph on silica gel to afford the desired products.
80%
With C21H17N3O; copper; potassium hydroxide In propan-1-ol at 80℃; for 5 h; Sealed tube
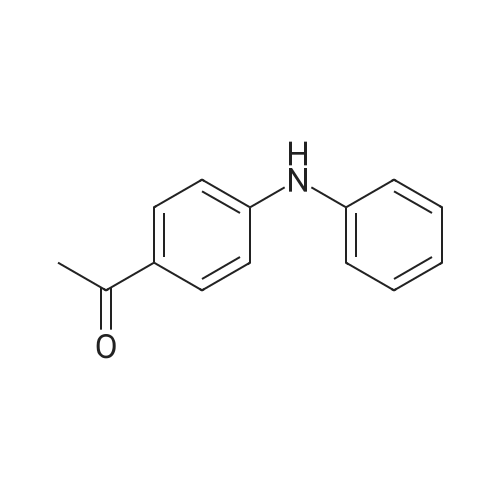
The 198 mg (1mmol) 4 - bromophenylmethyl, 279 mg (3mmol) aniline, 64 mg (1mmol) Cu, 65.4 mg (0.2mmol) ligand L6, 56mg (1mmol) KOH, 2 ml propanol, adding 10 ml reaction tube, sealing, 80 °C reaction under the condition of 5h. After the stop of the reaction, water, extracted with ethyl acetate, washing, saturated salt water washing, after drying with anhydrous sodium sulfate, filtered, the filtrate is distilled under reduced pressure, purification by silica gel chromatography separation column column, shall be 1 - (4 - (phenylamino) phenyl) b one 169 mg, yield 80percent.
75%
With C104H96N16O8Pd2(4+)*4NO3(1-); sodium t-butanolate In toluene at 110℃; for 18 h;
General procedure: In a 50 mL round bottom flask, the mixture of iodobenzene (2 mmol), amine (2.4 mmol), t-BuONa (3 mmol), and 1 as catalyst (0.05 mol percent) was taken in toluene (10 mL). The reaction mixture was then heated to 110°C and continued for 12–18 h. The progress of the reaction was monitored by TLC. Upon completion of the reaction the aqueous reaction mixture was extracted with ethyl acetate, washed with brine, dried over MgSO4, concentrated, and purified by column chromatography on silica gel which afforded corresponding coupling products (yield 75–96percent).
53%
at 90℃; for 12 h; Sealed tube
General procedure: To a 10 mL sealed vial was added Cu(OAc)2·H2O (10 mg, 0.05mmol), N-methoxy-1H-pyrrole-2-carboxamide (7 mg, 0.05 mmol), aryl bromide (1.0 mmol), amine (3.0 mmol), K3PO4 (318 mg, 1.5 mmol),PEG-100 (2.0 g) and a magnetic stir bar. The reaction mixture was stirred in an oil bath preheated to 90 °C for 12 h. After allowing the mixture to cool to room temperature, the reaction mixture was extracted with ethyl acetate (3 × 25 mL) and water (20 mL). The combined organic phases were washed with brine, dried over anhydrous Na2SO4, and concentrated in vacuo. The residue was purified by flash column chromatography on silica gel to afford the desired product.
82 %Chromat.
at 150℃; for 12 h;
General procedure: In a typical run, an oven-dried 10 ml round bottom flask was charged with a known mole percent of catalyst, NaOtBu (1.3 mmol), amine (1.2 mmol) and aryl halide (1 mmol) with the appropriate solvent(s) (4 ml). The flask was placed in a preheated oil bath at required temp. After the specified time the flask was removed from the oil bath, water (20 ml) was added, and extraction with ether (4×10 ml) was done. The combined organic layers were washed with water (3×10 ml), dried over anhydrous Na2SO4, and filtered. Solvent was removed under vacuum. The residue was dissolved in acetonitrile and analyzed by GC–MS.
Reference:
[1] Journal of Organometallic Chemistry, 2017, vol. 841, p. 57 - 61
[2] Chemical Communications, 1998, # 15, p. 1509 - 1510
[3] Applied Organometallic Chemistry, 2011, vol. 25, # 5, p. 341 - 347
[4] Synthesis (Germany), 2018, vol. 50, # 19, p. 3911 - 3920
[5] Tetrahedron, 2013, vol. 69, # 42, p. 8974 - 8977
[6] Patent: CN106883132, 2017, A, . Location in patent: Paragraph 0053-0055
[7] Applied Organometallic Chemistry, 2017, vol. 31, # 11,
[8] European Journal of Organic Chemistry, 2010, # 17, p. 3219 - 3223
[9] Tetrahedron Letters, 2016, vol. 57, # 14, p. 1532 - 1536
[10] European Journal of Organic Chemistry, 2009, # 5, p. 635 - 642
[11] Angewandte Chemie - International Edition, 2016, vol. 55, # 42, p. 13219 - 13223[12] Angew. Chem., 2016, vol. 128, # 42, p. 13413 - 13417,5
[13] Russian Chemical Bulletin, 2012, vol. 61, # 5, p. 1009 - 1013[14] Izv. Akad. Nauk, Ser. Khim., 2012, vol. 61, # 5, p. 1004 - 1008,5
[15] Synthesis (Germany), 2014, vol. 46, # 24, p. 3356 - 3364
[16] Russian Journal of General Chemistry, 2005, vol. 75, # 2, p. 207 - 211
[17] Inorganica Chimica Acta, 2019, vol. 486, p. 232 - 239
3
[ 99-90-1 ]
[ 23600-83-1 ]
Reference:
[1] Bulletin de la Societe Chimique de France, 1969, p. 2342 - 2355
4
[ 99-90-1 ]
[ 1000994-95-5 ]
Yield
Reaction Conditions
Operation in experiment
77%
With diethylamino-sulfur trifluoride In dichloromethane at 50℃; Inert atmosphere
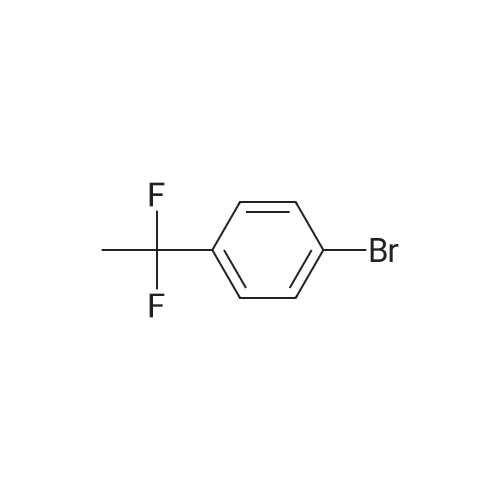
Example 63A1-Bromo-4-(1,1-difluoroethyl)benzene; Under argon, 3.0 g (15.07 mmol) of 1-(4-bromophenyl)ethanone were initially charged in 30 ml of dichloromethane, and 15.9 ml (120.57 mmol) of [ethyl(trifluoro-λ4-sulphanyl)amino]ethane (DAST) were added slowly. The reaction solution was then slowly warmed to 50° C. and stirred at this temperature overnight. After the reaction had ended, the reaction solution was slowly poured into ice-water. The organic phase was then separated off, and the aqueous phase was extracted three more times with dichloromethane. The combined organic phases were dried over magnesium sulphate. After filtration, the solvent was removed under reduced pressure. The crude product was purified chromatographically on silica gel (mobile phase petroleum ether/dichloromethane 4:1). This gave 2.56 g (11.58 mmol, 77percent of theory) of the title compound as a yellowish liquid.GC-MS (Method 1): Rt=2.84 min; m/z=220/222 (M)+.
76%
With (bis-(2-methoxyethyl)amino)sulfur trufluoride In tetrahydrofuran; methanol for 96 h; Reflux
Example 48A 1-Bromo-4-(1,1-difluoroethyl)benzene A solution of 10.0 g (50.2 mmol) of 4-bromoacetophenone in tetrahydrofuran (20 ml) was admixed with 50.0 ml (151 mmol, 50percent in tetrahydrofuran) of bis(2-methoxyethyl)aminosulphur trifluoride (Deoxofluor) and 3 drops of methanol, and then stirred under reflux for 4 days. The reaction mixture was cautiously added dropwise to a mixture of saturated aqueous sodium hydrogencarbonate solution and ice (1:1) and then extracted with diethyl ether. The organic phase was dried over sodium sulphate, filtered and concentrated under reduced pressure. The residue was purified by means of column chromatography (silica gel, petroleum ether/dichloromethane 3:1). Yield: 8.46 g (76percent of theory) 1H NMR (400 MHz, DMSO-d6): δ=7.70 (d, 2H), 7.52 (d, 2H), 1.96 (t, 3H).
76%
With methanol; (bis-(2-methoxyethyl)amino)sulfur trufluoride In tetrahydrofuran for 96 h; Reflux
Example 68A1-Bromo-4-(1,1-difluoroethyl)benzene A solution of 10.0 g (50.2 mmol) of 4-bromoacetophenone in tetrahydrofuran (20 ml) was admixed with 50.0 ml (151 mmol, 50percent in tetrahydrofuran) of bis(2-methoxyethyl)aminosulphur trifluoride (Deoxofluor) and 3 drops of methanol, and then stirred under reflux for four days. The reaction mixture was cautiously added dropwise to a mixture of saturated aqueous sodium hydrogencarbonate solution and ice (1:1) and then extracted with diethyl ether. The organic phase was dried over sodium sulphate, filtered and concentrated under reduced pressure. The residue was purified by means of column chromatography (silica gel, petroleum ether/dichloromethane 3:1). Yield: 8.46 g (76percent of theory)1H NMR (400 MHz, DMSO-d6): δ=7.70 (d, 2H), 7.52 (d, 2H), 1.96 (t, 3H).
59%
at 85℃; for 15 h;
Deoxo-Fluor (registered trademark) (22.2 g) was added to 1-(4-bromophenyl)ethanone (20.0 g) and the mixturewas stirred at 85°C for 15 hours. Under icecooling, ice water and an aqueous solution of potassium carbonate wereadded to the reaction solution, followed by extraction with chloroform. The solvent was evaporated under reducedpressure and the obtained residue was purified by silica gel column chromatography (hexane) to give the title compound(13.0 g, yield 59percent) as a yellow oil.1H NMR (600 MHz, CDCl3) δ ppm 1.91 (t, J=18.2 Hz, 3H), 7.50 (d, J=8.3 Hz, 2H), 7.86 (d, J=8.3 Hz, 2H).
25%
With (bis-(2-methoxyethyl)amino)sulfur trufluoride In dichloromethane; toluene at 20 - 85℃; for 20 h; Ionic liquid; Sealed tube
Step A: Preparation of l-bromo-4-(l,l-difluoroethyl)benzene. [00211] To a vial equipped with a magnetic stir bar were added a solution of l-(4- bromophenyl)ethanone (295 mg, 1.48 mmol) in anhydrous DCM (3.0 mL) followed by a 50percent solution of Deoxofluor? in toluene (1.6 mL, 4.45 mmol) at room temperature under N2 and the vial was sealed. The reaction mixture was stirred for approximately 15 h at room temperature, but little conversion had taken place. The mixture was concentrated, treated with additional Deoxofluor? solution (0.66 mL, 1.79 mmol),and warmed to and stirred at 85 °C under N2 for 5 h. The reaction mixture was cooled to 0 °C and carefully quenched by adding sat'd aq NaHC03 dropwise until gas evolution ceased. The bi-phasic mixture was extracted with DCM (2 x 5 mL), and the combined extracts were dried over Na2S04, filtered, and concentrated. The residue was purified by column chromatography (Si02, 0-^50 EtOAc in hexanes) to afford the title compound (83 mg, 25percent)as a clear liquid: 1H NMR (400 MHz, CDC13) δ 7.58 - 7.53 (m, 2H), 7.41 - 7.35 (m, 2H), 1.90 (t, J = 18.1 Hz, 3H); 19F NMR (376 MHz, CDC13) δ -87.86; IR (Thin Film) 1599, 1294, 1089 cm"1; EIMS mlz 220/221.
17.26 g
With (bis-(2-methoxyethyl)amino)sulfur trufluoride In chloroform at 50℃; for 35 h; Inert atmosphere
Under argon atmosphere, into a 500-ml reaction vessel made of tetrafluoroethylene-perfluoroalkyl vinyl ether copolymer (PFA) and equipped with a stirring apparatus were placed 25 g (126 mmol) of 4-bromoacetophenone, 111 g (500 mmol) of bis(2-methoxyethyl)aminosulfur trifluoride, and 250 ml of anhydrous chloroform, so that a homogeneous solution was prepared. And then, the solution was reacted at an internal temperature of about 50°C for 35 hours. Subsequently, the reaction solution was cooled to room temperature, and then the reaction solution was added to 1000 ml of a saturated aqueous solution of sodium hydrogen carbonate, which was cooled in ice. Subsequently, the mixture was subjected to extraction with 500 ml of chloroform. The solvent was distilled off under a reduced pressure, and then the reaction mixture was purified by silica gel column chromatography (hexane: 100 vol percent), to provide 17.26 g of Compound (4-1) in the form of a colorless liquid. [0134] The properties of Compound (4-1) were as follows. 1H-NMR (400MHz, CDCl3, 8 (ppm)); 1.90 (3H, t, J=18.1Hz), 7.54 (2H, d, J=2.3Hz), 7.57 (2H, d, J=2.4Hz) CI-MS; 222 (M+2)
17.26 g
With (bis-(2-methoxyethyl)amino)sulfur trufluoride In chloroform at 50℃; for 35 h; Inert atmosphere
Under argon atmosphere, into a 500-ml reaction vessel 30 made of tetrafluoroethylene-perfluoroalkyl vinyl ether copolymer (PFA) and equipped with a stirring apparatus were placed 25 g (126 mmol) of 4-bromoacetophenone, 111 g (500 mmol) of bis(2-methoxyethyl)aminosulthr trifluoride, and 250 ml of anhydrous chloroform, so that a homogeneous solution was prepared. And then, the solution was reacted at an internal temperature of about 50° C. for 35 hours. Subsequently, the reaction solution was cooled to room temperature, and then the reaction solution was added to 1000 ml of a saturated aqueous solution of sodium hydrogen carbonate, which was cooled in ice. Subsequently, the mixture was subjected to extraction with 500 ml of chloroform. The solvent was distilled off under a reduced pressure, and then the reaction mixture was purified by silica gel column chromatography (hexane: 100 vol percent), to provide 17.26 g of Compound (4-1) in the form of a colorless liquid.The properties of Compound (4-1) were as follows. ‘H-NMR (400 MHz, CDC13, ? (ppm)); 1.90 (3H, t, J=18. 1 Hz), 7.54 (2H, d, J=2.3 Hz), 7.57 (2H, d, J=2.4 Hz)CI-MS; 222 (M+2)4045
palladium diacetate; In tetrahydrofuran; nitrogen; toluene;
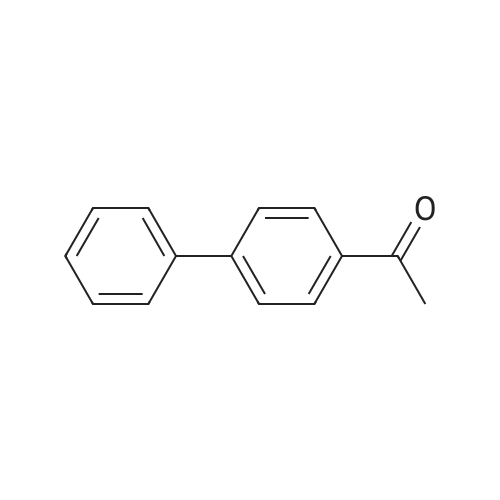
EXAMPLE 45 Synthesis of 4-acetylbiphenyl from an aryl bromoide utilizing 0.000001 mol percent Pd An oven dried resealable Schlenk tube was evacuated and backfilled with argon and charged with phenylboronic acid (228 mg, 1.5 mmol), and potassium phosphate (425 mg, 2.0 mmol), and 4-bromoacetophenone (199 mg, 1.0 mmol). The tube was evacuated and backfilled with argon, and toluene (1.5 mL) was added through a rubber septum. In a separate flask in a nitrogen filled glovebox, palladium acetate (4.5 mg, 0.02 mmol) and 2-(di-tert-butylphosphino)biphenyl (12.0 mg, 0.04 mmol) were dissolved in 20 ml, THF under argon. A portion of this solution (10 muL, 0.00001 mmol Pd, 0.001 mol percent Pd) was added to a second flask contaninig 10 mL THF). A portion of this second solution (10 muL, 0.00000001 mmol Pd, 0.000001 mol percent Pd) was added to the reaction mixture, followed by additional toluene (1.5 mL) through a rubber septum. The tube was sealed with a teflon screwcap, and the reaction mixture was heated to 100° C. with stirring until the starting aryl bromide had been completely consumed as judged by GC analysis. The reaction mixture was then diluted with ether (30 mL) and poured into a separatory funnel. The mixture was washed with water (20 mL), and the aqueous layer was extracted with ether (20 mL). The combined organic layers were washed with brine (20 mL), dried over anhydrous magnesium sulfate, filtered, and concentrated. The crude material was purified by flash chromatography on silica gel to afford 176 mg (90percent) of the title compound.
176 mg (90%)
palladium diacetate; In tetrahydrofuran; nitrogen; toluene;
Example 45 Synthesis of 4-acetylbiphenyl from an aryl bromoide Utilizing 0.000001 mol percent Pd An oven dried resealable Schlenk tube was evacuated and backfilled with argon and charged with phenylboronic acid (228 mg, 1.5 mmol), and potassium phosphate (425 mg, 2.0 mmol), and 4-bromoacetophenone (199 mg, 1.0 mmol). The tube was evacuated and backfilled with argon, and toluene (1.5 mL) was added through a rubber septum. In a separate flask in a nitrogen filled glovebox, palladium acetate (4.5 mg, 0.02 mmol) and 2-(di-tert-butylphosphino)biphenyl (12.0 mg, 0.04 mmol) were dissolved in 20 mL THF under argon. A portion of this solution (10 muL, 0.00001 mmol Pd, 0.001 mol percent Pd) was added to a second flask contaninig 10 mL THF). A portion of this second solution (10 muL, 0.00000001 mmol Pd, 0.000001 mol percent Pd) was added to the reaction mixture, followed by additional toluene (1.5 mL) through a rubber septum. The tube was sealed with a teflon screwcap, and the reaction mixture was heated to 100° C. with stirring until the starting aryl bromide had been completely consumed as judged by GC analysis. The reaction mixture was then diluted with ether (30 mL) and poured into a separatory funnel. The mixture was washed with water (20 mL), and the aqueous layer was extracted with ether (20 mL). The combined organic layers were washed with brine (20 mL), dried over anhydrous magnesium sulfate, filtered, and concentrated. The crude material was purified by flash chromatography on silica gel to afford 176 mg (90percent) of the title compound.
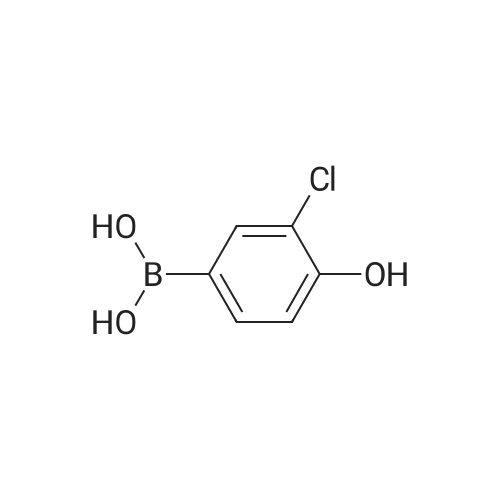
With tetrabutylammomium bromide; potassium carbonate;palladium diacetate; In ethanol; water; at 150℃; for 0.166667h;Irradiation;
l-(3'-ChIoro-4'-hydroxy-biphenyl-4-yl)-ethanone 84. To a mixture of 4'-bromoacetophenone (0.10Og, 0.5 mmol), 3-chloro-4- hydroxyphenylboronic acid 83 (0.103 g, 0.6 mmol), K2CO3 (0.173 g5 1.25 mmol) and Bu4NBr (0.161 g, 0.5 mmol) in EtOH (1.2 mL) and water (2.8 mL) was added Pd(OAc)2 (catalytic) and the reaction was micro waved at 15O0C for 10 min. Water (10 mL) was added and the organics extracted into EtOAc (10 mL). Flash chromatography using an elution gradient of hexane to 30percent EtOAc in hexane gave the product in a mixture. This was dissolved in EtOAc and extracted with base. The aqueous layer was acidified and organics extracted into EtOAc then concentrated under reduced pressure. Flash chromatography using DCM as eluent gave the product as the first fraction: 1H NMR delta (270 MHz5 CDCl3) 2.62 (3H, s), 5.86 (IH5 s), 7.11 (IH5 d5 J= 8.4 Hz), 7.44 (IH, dd, J= 8.4, 2.2 Hz), 7.57-7.62 (3H, m), 8.00 (2H5 d, J= 8.7 Hz); HPLC > 92percent (Rt 3.98, 90percent MeCN in H2O); ES-ve MS (M-H)- 245.22 m/z
naphthalen-1-yl-acetic acid [1-(4-bromophenyl)ethylidene]hydrazide[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
60.3%
In ethanol;Reflux;
General procedure: solutionof acid hydrazide (0.01 mol) and appropriate benzaldehyde/acetophenone (0.01 mol) in ethanol was refluxed for 5-6 h. The precipitated title compounds were then filtered off, washed with water and recrystallized from ethanol.






 Chemistry
Chemistry
 Pharmaceutical Intermediates
Pharmaceutical Intermediates
 Inhibitors/Agonists
Inhibitors/Agonists
 Material Science
Material Science







 For Research Only
For Research Only
 120K+ Compounds
120K+ Compounds
 Competitive Price
Competitive Price
 1-2 Day Shipping
1-2 Day Shipping