* All experimental methods are cited from the reference, please refer to the original source for details. We do not guarantee the accuracy of the content in the reference.
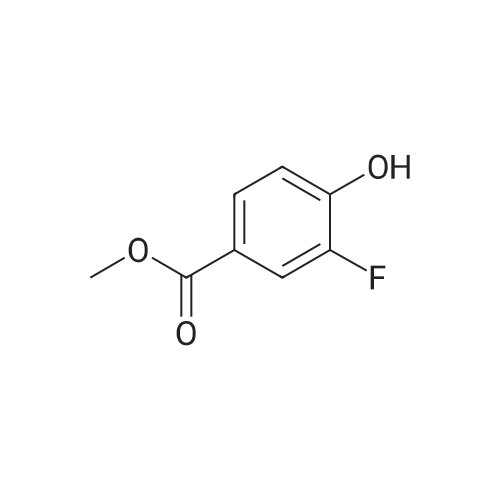
With sodium percarbonate; acetic acid; potassium iodide; In water; for 2.5h;
General procedure: Potassium iodide (2.1 equiv) was added with stirring to 50% (v/v) aq. AcOH (1.0 M) containing a phenol derivative S5, and sodium percarbonate (SPC) (3.8 equiv) was slowly added over 30 min. The stirring was continued for 2.0 h at 50 oC under a reflux condenser. After cooling, the reaction mixture was slowly poured into a vigorously stirred biphasic mixtures made of CH2Cl2 and saturated aqueous Na2SO3. Separated aqueous layer was extracted twice with CH2Cl2. The combined organic layer was washed with saturated aqueous NaCl. The organic layer was dried over anhydrous Na2SO4 and the solvent was removed under reduced pressure. The crude product was purified by column chromatography to provide 23.
99%
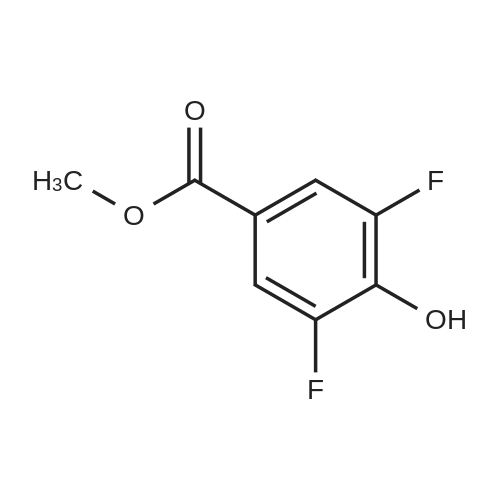
With acetic acid; periodic acid; potassium iodide; In water; at 60℃;
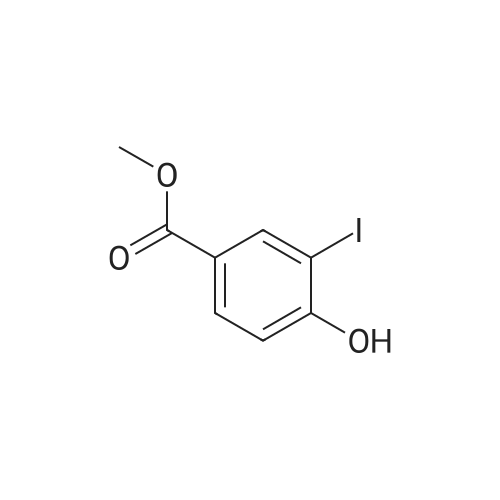
30.4 g of methyl p-hydroxybenzoate was added to a 2 L single-necked flask, 1 L of acetic acid, 500 ml of water, 42.8 g of periodic acid and 66.4 g of potassium iodide were added and reacted at 60 C overnight.The TLC plate showed complete reaction, where the TLC developing agent was PE: EA = 5: 1.The reaction solution was cooled to room temperature, added to 2 L of water, filtered, and the solid was dried with an infrared lamp to obtain 80 g of a yellow solid, that is, a compound having a structure represented by the formula M2-1 in a yield of 99%.
98.8%
10 g (65.7 mmol) of p-methoxybenzoate , 80 ml of methanol were added thereto, stirred and dissolved, and 11.3 g of sodium acetate and 7 g of water were added thereto, stirred and dissolved, and 30 g of iodine (118.3 mmo 1) was added thereto, and the temperature was raised to 70 C. After reacting for 2 hours, a sodium hydroxide solution (sodium hydroxide 5.5 g (137.5 mmol) and water 200 ml) was added, and the reaction was kept at 70 C for 2 hours, and the temperature was slowly lowered to room temperature, and 10 g of a 25% sodium hydrogen sulfite solution was added dropwise thereto. The color of the liquid was removed, and the crystals were further stirred for 1 hour, filtered, and the wet product was dried by hot air at 50 for 8 hours to obtain 26.2 g of a white crystalline powder, yield 98.8%.
86.7%
To methanol (150 mL)Was added methyl p-hydroxybenzoate (10 g, 65.7 mmol)Sodium acetate hydrate (18.78 g, 138 mmol) andIodine (35 g, 137.9 mmol)The resulting mixture was stirred under reflux for 1.5 hours.A solution of sodium hydroxide (5.52 g, 138 mmol)Of water (200 mL)Then reflux for 2.5 hours.Cool to room temperature and add dilute sulfurSodium hydrogen acid solution to the color faded. The filter cake was washed with a small amount of water and then dissolved with ethyl acetate (200 mL), anhydrous sulfuric acidSodium dry. The solvent was evaporated under reduced pressure and the resulting product was recrystallized from petroleum ether / ethyl acetate,4-hydroxy-3,5-diiodobenzoate (11) (23 g). The yield was 86.7%.
With potassium carbonate; In acetonitrile; at 60℃; for 5h;
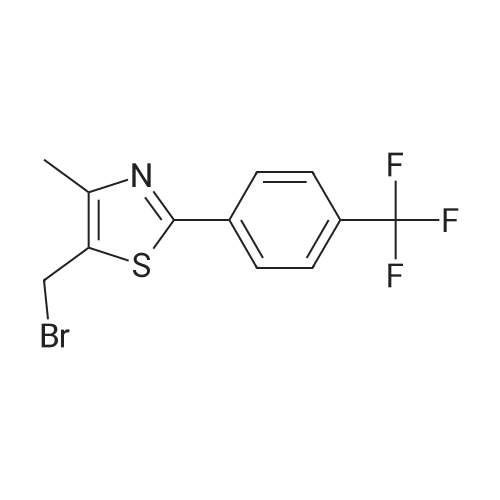
Example 6; Intermediate: 4-14-Methyl-2-(4-trifluoromethvl-phenvl)-thiazol-5-ylmethoxyl-benzoic acid methyl ester; To a mixture of 4-hydroxy-benzoic acid methyl ester (152 mg, 1 mmol) and potassium carbonate (152 mg, 1.1 mmol) add acetonitrile (4 mL). To this mixture add 5-bromomethyl-4- methyl-2- (4-trifluoromethyl-phenyl)-thiazole (Example 4, 314 mg, 1 mmol). Warm the resulting reaction mixture to 60°C, and stir at this temperature for 5 hrs. Cool the mixture to room temperature, dilute with ethyl acetate, wash with water, then brine, dry over MgS04, filter and concentrate to give the title compound as a white solid (364 mg). MS (ESI) m/z 408 (M+H) ; H1 NMR (CDCl3) 8 2.53 (s, 3H), 3.90 (s, 3H), 4.52 (s, 2H), 7.00 (d, J = 9Hz, 2H), 7.68 (d, J = 8Hz, 2H), 8.02 (m, 4H).
With triphenylphosphine;SiO2; In methanol; dichloromethane;
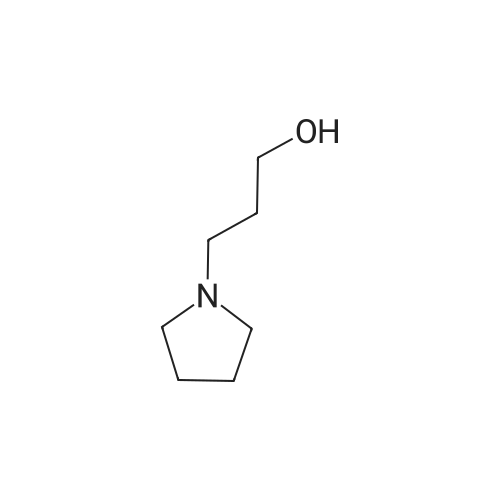
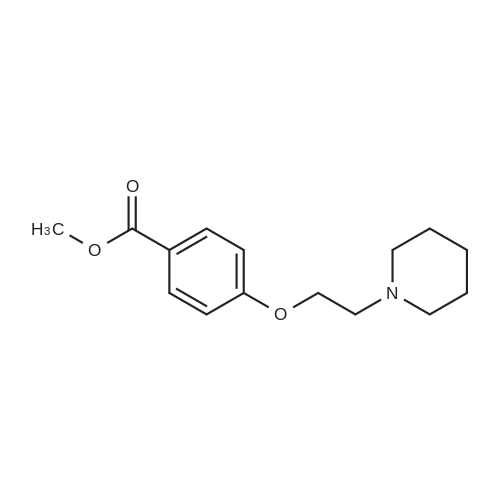
Part B. Methyl 4-[3-(1-Pyrrolidinyl)propoxy]benzoate STR40 A solution of 6.25 g (23.8 mmol) of triphenylphosphine, 3.30 g (21.7 mmol) of methyl 4-hydroxybenzoate, and 2.80 g (21.7 mmol) of 1-(3-hydroxypropyl)pyrrolidine in 100 mL of CH2 Cl2 was treated with 3.80 mL (24.1 mmol) of diethyl azodicarboxylate in a dropwise manner. The reaction was stirred at ambient temperature for 16 h and was quenched by the addition of 20 mL of brine. The two layers were separated, and the organic layer was dried over K2 CO3 and concentrated to give 6.10 g of an oily solid which was purified by flash chromatography (SiO2; 0-5percent MeOH in CH2 Cl2) to afford 2.46 g (9.34 mmol; 43percent) of the desired product. FDMS 263 (M+; 100); HRMS Calcd for C15 H21 NO3: 264.1600. Found: 264.1609.
With potassium carbonate; In Isopropyl acetate; water;
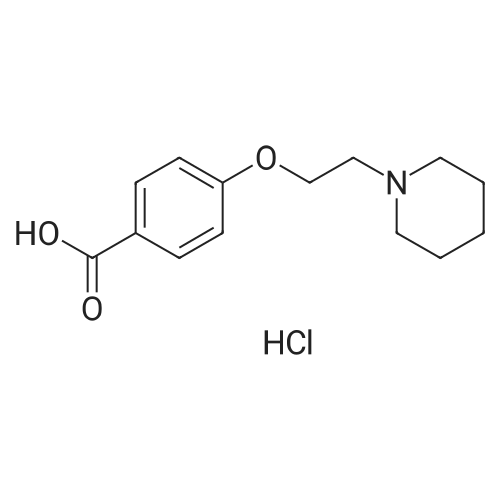
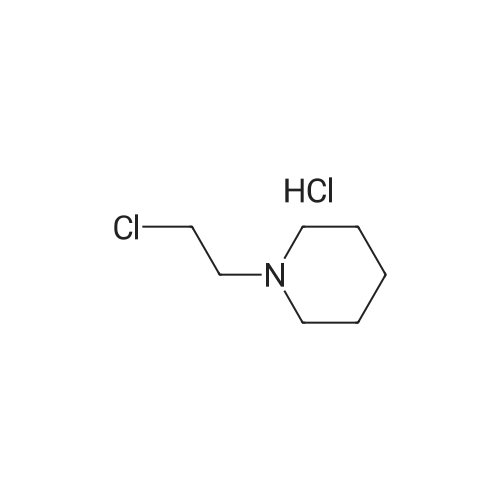
EXAMPLE 4 Preparation of 4-(2-piperidinoethoxy)benzoic acid hydrochloride To a 250 mL 3 neck flask equipped with mechanical stirring, condenser, and heating apparatus consisting of a RTD probe hooked via a temperature controller to a heating mantle and under nitrogen atmosphere, the following were added: 7.61 g methyl 4-hydroxybenzoate, 11.05 g beta-chloroethylpiperidine hydrochloride, 16.59 g powdered potassium carbonate, and 60 mL isopropyl acetate. The mixture was heated slowly to 80° C. After 5 hours, high performance liquid chromatography showed reaction to be 90percent complete. After being left overnight at 80° C., reaction was complete. The mixture was then cooled to ambient temperature, after which 60 mL deionized water was added. The mixture was stirred until all solids dissolved. The aqueous layer was separated and discarded. The organic layer was extracted 3 times with 20 mL 4N hydrochloride. The combined aliquots, containing 4-(2-piperidinoethoxy)benzoic acid, ethyl ester, were heated at reflux (92° C., 30 minutes required to reach reflux). After 7.5 hours at reflux, the mixture was then distilled to remove approximately 10 mL water and cooled in an ice bath for 15 minutes. The resulting crystalline 4-(2-piperidinoethoxy)benzoic acid, hydrochloride was removed by filtration and rinsed with acetone and dried. Yield=12.53 g of product (87.7percent of theoretical).
With potassium carbonate; In water; ethyl acetate;
EXAMPLE 9 Preparatlon of 4-(2-piperidinoethoxy)benzoic acid hydrochloride To a 250 mL 3 neck flask with mechanical stirring and condenser, and a heating apparatus consisting of an RTD probe in the flask hooked via a temperature controller to a heating mantle and under nitrogen atmosphere, the following was added: 7.61 g of methyl 4-hydroxybenzoate, 11.05 g of beta-chloroethylpiperidine hydrochloride, 16.59 g of powdered potassium carbonate and 60 mL of ethyl acetate. The mixture was heated slowly to reflux. After overnight reflux, the mixture was cooled to ambient temperature, after which 60 mL of deionized water was added. The aqueous layer was separated and discarded. The organic layer was extracted with 4N hydrochloride (3 aliquots of 20 mL). The combined acid extracts were heated to reflux. After 1 hour at reflux, HPLC indicated the saponification to be 60percent complete. After 4 hours, the reaction was near 100percent complete. The mixture was cooled to 0° C.-5° C. and stirred. The resulting crystals were filtered, rinsed with acetone and dried.
With potassium carbonate; In Isopropyl acetate; water;
a. To a 250 mL 3 neck flask, with mechanical stirring, condenser, and RTD probe were added the following under nitrogen atmosphere: 0.05 mol methyl 4-hydroxybenzoate, 0.06 mol beta-chloroethylpiperidine hydrochloride, 16.59 grams of potassium carbonate, and 60 mL of isopropyl acetate. The mixture was heated at 75° C.-80° C. for 20 hours, at which time all the methyl 4-hydroxybenzoate was consumed. 60 mL of water was then added to dissolve the potassium carbonate. The organic and aqueous phases were then Separated and the aqueous layer discarded. The organic layer was washed with a second 60 mL aliquot of water; the layers were separated and the aqueous layer discarded. The reaction product, 4-(2-piperidinoethoxy)benzoic acid, methyl ester, was then extracted into 25 mL 8N hydrochloric acid. The aqueous phase was separated and the organic phase discarded. The aqueous phase was refluxed in a 50 mL round bottomed flask with magnetic stirring and condenser for 48 hours. The mixture was then cooled to 0° C.-5° C. and the crystals removed by filtration. The crystals were rinsed with acetone and dried overnight in 50° C. vacuum oven. 13.63 g of 4-(2-piperidinoethoxy)benzoic acid hydrochloride were recovered, which is 95.3percent of the theoretical yield.
With hydrogenchloride; potassium carbonate; In water;
EXAMPLE 5 Preparation of 4-(2-piperidinoethoxy)benzoic acid hydrochloride To a 125 mL 3 neck flask with mechanical stirring, condenser, and a heating apparatus consisting of an RTD probe hooked via a temperature controller to a heating mantle, the following were added: 7.61 g methyl 4-hydroxybenzoate, 11.05 g beta-chloroethylpiperidine hydrochloride, 16.59 g powdered potassium carbonate, and 60 mL amyl acetate. The mixture was heated in an oil bath under nitrogen to 115° C.-120° C. for 4 hour. HPLC indicated that the reaction was complete. The mixture was then cooled to ambient temperature and 40 mL of deionized water were added to dissolve solids. The aqueous layer was separated and discarded and the water wash was repeated. 5 mL of the organic phase was removed as an analytical standard. 25 mL 8N hydrochloric acid was added to remaining organic phase to extract the intermediate. The layers were separated and the acidified aqueous layers returned to the reaction flasks. The organic phase was discarded. The aqueous phase was heated to 95° C. until HPLC indicated complete hydrolysis of the ester (about 4 hours). The mixtures were cooled to 0° C.-5° C. for 1 hour and filtered. The filter cakes were rinsed with acetone (approx. 25 mL) and dried. Yield 12.61 g (83.6percent theoretical).
With hydrogenchloride; potassium carbonate; In water; acetone;
EXAMPLE 7 Preparation of 4-(2-piperidinoethoxy)benzoic acid hydrochloride To a 125 mL 3 neck flask with mechanical stirring, condenser, and a heating apparatus consisting of an RTD probe in the flask hooked via a temperature controller to a heating mantle, the following were added: 7.61 g methyl 4-hydroxybenzoate, 11.05 g beta-chloroethylpiperidine hydrochloride, 16.59 g powdered potassium carbonate, and 60 mL amyl acetate. The mixture was heated overnight under nitrogen in an oil bath to 125° C., and was allowed to proceed until HPLC indicated complete consumption of the methyl 4-hydroxybenzoate. The mixture was cooled to ambient temperature and 40 mL deionized water was added to dissolve the solids. The aqueous layer was separated and discarded. The water wash was repeated. 25 mL of 8N hydrochloric acid were added to extract intermediate. The layers were separated and the acid layer returned to the reaction flask. The acid solution was heated to 95° C. for about 24 hours as a "stress" test; (after 6 hours 1percent of the uncleaved ester remained.) The mixture was cooled to 40° C. and 25 mL acetone added. The mixture was cooled to 0° C.-5° C. for 1 hour. The mixture was filtered and the cakes rinsed with approx. 25 mL acetone and dried. Yield=12.0 g (84.1percent of theoretical).
With potassium carbonate; In dichloromethane; acetone;
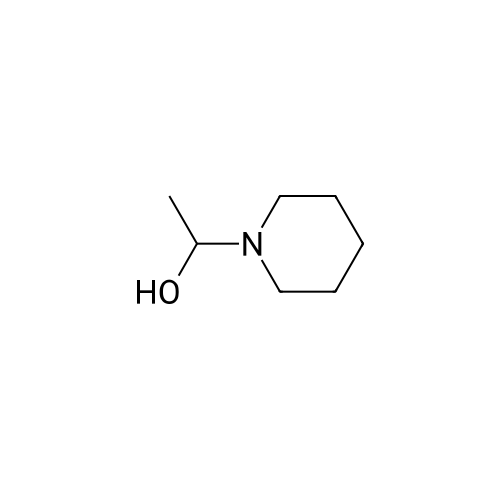
Preparation 2 4-(2-piperidinoethoxy) benzoic acid hydrochloride Into a 50 ml round bottom flask is placed 6.40 g (33.6 mmol) of p-toluenesulfonyl chloride and 25 ml of methylene chloride. The resulting solution is cooled with an ice bath as 4.00 g (31.0 mmol) of 1-piperidinoethanol in 6 ml of methylene chloride is added dropwise. After the addition is complete, the ice bath is removed and the resulting slurry is stirred for about 12 hours. The reaction mixture is concentrated on a rotary evaporator to yield a solid residue. The solid residue is transferred to a 100 ml round bottom flask with 45 ml of amyl acetate. Potassium carbonate (6.87 g, 49.7 mmol) and methyl 4-hydroxybenzoate (2.82 g, 18.5 mmol) are added to the slurry. The resulting mixture is heated to 145° C. for 2 hours. The reaction mixture is cooled to room temperature and washed twice with water. The organic layer is extracted with 11 ml of 8N hydrochloric acid. The aqueous layer is heated at gentle reflux for 3 hours. The resulting slurry is cooled to 50° C. and 11 ml of acetone is added. The slurry is cooled to 0°-5° C. and stirred for 1 hour. The product is filtered, washed with cold acetone, and dried at 50° C. in vacuo to yield 920 mg of product, (17percent).
With potassium carbonate; In dichloromethane; acetone;
Example 2 Preparation of 4-(2-piperidinoethoxy) benzoic acid hydrochloride Into a 50 ml round bottom flask is placed 6.40 g (33.6 mmol) of p-toluenesulfonyl chloride and 25 ml of methylene chloride. The resulting solution is cooled with an ice bath as 4.00 g (31.0 mmol) of 1-piperidinoethanol in 6 ml of methylene chloride is added dropwise. After the addition is complete, the ice bath is removed and the resulting slurry is stirred for about 12 hours. The reaction mixture is concentrated on a rotary evaporator to yield a solid residue. The solid residue is transferred to a 100 ml round bottom flask with 45 ml of amyl acetate. Potassium carbonate (6.87 g, 49.7 mmol) and methyl 4-hydroxybenzoate (2.82 g, 18.5 mmol) are added to the slurry. The resulting mixture is heated to 145°C for 2 hours. The reaction mixture is cooled to room temperature and washed twice with water. The organic layer is extracted with 11 ml of 8 N hydrochloric acid. The aqueous layer is heated at gentle reflux for 3 hours. The resulting slurry is cooled to 50°C and 11 ml of acetone is added. The slurry is cooled to 0-5°C and stirred for 1 hour. The product is filtered, washed with cold acetone, and dried at 50°C in vacuoto yield 920 mg of product, (17percent).
With potassium carbonate; In dichloromethane; 1-pentyl acetate; acetone;
Preparation 3 4-(2-piperidinoethoxy) benzoic acid hydrochloride Into a 50 ml round bottom flask is placed 4.93 g (27.0 mmol) of 2,2,2-trifluoroethanesulfonyl chloride and 25 ml of methylene chloride. The resulting solution is cooled with an ice bath as 3.22 g (24.9 mmol) of 1-piperidinoethanol in 7 ml of methylene chloride is added dropwise. After the addition is complete, the ice bath is removed and the resulting slurry is stirred for about 12 hours. The reaction mixture is concentrated on a rotary evaporator to yield a waxy residue. The solid residue is slurried in 45 ml of amyl acetate and then potassium carbonate (5.50 g, 39.9 mmol) and methyl 4-hydroxybenzoate (2.26 g, 14.8 mmol) are added. The resulting mixture is heated to 140° C. for 2 hours. The reaction mixture is cooled to room temperature and washed twice with water and the organic layer is extracted with 10.5 ml of 8N hydrochloric acid. The aqueous layer is heated at gentle reflux for 4 hours. The resulting slurry is cooled to 50° C. and 11 ml of acetone is added. The slurry is cooled to 0°-5° C. and stirred for 1 hour. The product is filtered, washed with cold acetone, and dried at 50° C. in vacuo to yield 3.94 g of product, (90percent).
With potassium carbonate; In dichloromethane; 1-pentyl acetate; acetone;
Example 3 Preparation of 4-(2-piperidinoethoxy) benzoic acid hydrochloride Into a 50 ml round bottom flask is placed 4.93 g (27.0 mmol) of 2,2,2-trifluoroethanesulfonyl chloride and 25 ml of methylene chloride. The resulting solution is cooled with an ice bath as 3.22 g (24.9 mmol) of 1-piperidinoethanol in 7 ml of methylene chloride is added dropwise. After the addition is complete, the ice bath is removed and the resulting slurry is stirred for about 12 hours. The reaction mixture is concentrated on a rotary evaporator to yield a waxy residue. The solid residue is slurried in 45 ml of amyl acetate and then potassium carbonate (5.50 g, 39.9 mmol) and methyl 4-hydroxybenzoate (2.26 g, 14.8 mmol) are added. The resulting mixture is heated to 140°C for 2 hours. The reaction mixture is cooled to room temperature and washed twice with water and the organic layer is extracted with 10.5 ml of 8 N hydrochloric acid. The aqueous layer is heated at gentle reflux for 4 hours. The resulting slurry is cooled to 50°C and 11 ml of acetone is added. The slurry is cooled to 0-5°C and stirred for 1 hour. The product is filtered, washed with cold acetone, and dried at 50°C in vacuoto yield 3.94 g of product, (90percent).
With thionyl chloride; potassium carbonate; In dichloromethane; water; acetone;
Preparation 1 4-(2-piperidinoethoxy) benzoic acid hydrochloride Into a 500 ml round bottom flask is placed 23.38 g (197 mmol) of thionyl chloride and 140 ml of methylene chloride. The resulting solution is cooled with an ice bath as 21.91 g (170 mmol) of 1-piperidinoethanol in 30 ml of methylene chloride is added over 30 minutes. After the addition is complete, the ice bath is removed and the mixture is stirred for about 12 hours. The reaction mixture is concentrated on a rotary evaporator to yield a solid residue. Amyl acetate (225 ml), potassium carbonate (34.4 g, 249 mmol), and methyl 4-hydroxybenzoate (14.1 g, 92 mmol) are added to the residue. The resulting slurry is heated to 135° C. for 5 hours. The reaction mixture is cooled to room temperature and washed twice with 100 ml of water. The organic layer is extracted with 53 ml of 8N hydrochloric acid. The aqueous layer is heated at gentle reflux for 4 hours. The resulting slurry is cooled to 50° C. and 50 ml of acetone is added. The slurry is cooled to 0°-5° C. and stirred for 1 hour. The product is filtered, washed with cold acetone, and dried at 50° C. in vacuo to yield 17.9 g of product, (67percent).
With thionyl chloride; potassium carbonate; In dichloromethane; water; acetone;
Example 1 Preparation of 4-(2-piperidinoethoxy)benzoic acid hydrochloride Into a 500 ml round bottom flask is placed 23.38 g (197 mmol) of thionyl chloride and 140 ml of methylene chloride. The resulting solution is cooled with an ice bath as 21.91 g (170 mmol) of 1-piperidinoethanol in 30 ml of methylene chloride is added over 30 minutes. After the addition is complete, the ice bath is removed and the mixture is stirred for about 12 hours. The reaction mixture is concentrated on a rotary evaporator to yield a solid residue. Amyl acetate (225 ml), potassium carbonate (34.4 g, 249 mmol), and methyl 4-hydroxybenzoate (14.1 g, 92 mmol) are added to the residue. The resulting slurry is heated to 135°C for 5 hours. The reaction mixture is cooled to room temperature and washed twice with 100 ml of water. The organic layer is extracted with 53 ml of 8 N hydrochloric acid. The aqueous layer is heated at gentle reflux for 4 hours. The resulting slurry is cooled to 50°C and 50 ml of acetone is added. The slurry is cooled to 0-5°C and stirred for 1 hour. The product is filtered, washed with cold acetone, and dried at 50°C in vacuoto yield 17.9 g of product, (67percent).
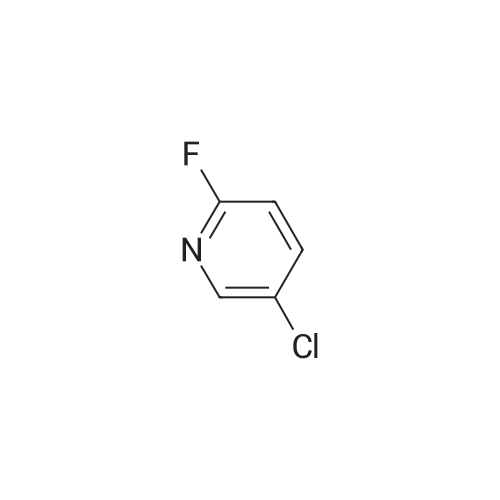
methyl 4-((5-chloropyridin-2-yl)oxy)benzoate[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
9 g
With caesium carbonate; In N,N-dimethyl-formamide; at 110℃; for 12h;
[0064] Methyl 4-hydroxybenzoate (6.5 g, 49 mmol), <strong>[1480-65-5]5-chloro-2-fluoropyridine</strong> (5.0 g, 33 mmol) and cesium carbonate(20 g, 65 mmol) were dissolved in N,N-dimethyl foramide (50 mL). The reaction solution was refluxed at 110 °C for 12hours, and the completion of reaction was monitored by thin layer chromatography (petroleum ether:ethyl acetate = 5:1),the solvent was rotary dried. The crude compound was partitioned between ethyl acetate (250 mL) and water (250 mL).The organic phase was dried over anhydrous sodium sulfate, filtered and evaporated under a reduced pressure. Thecrude compound was purified and separated by column chromatography to yield the product methyl 4-((5-chloropyridin-2-yl)oxy)benzoate (9.0 g, 93percent).MS: m/z 264.2 [M+1]






 Chemistry
Chemistry
 Pharmaceutical Intermediates
Pharmaceutical Intermediates
 Inhibitors/Agonists
Inhibitors/Agonists
 Material Science
Material Science







 For Research Only
For Research Only
 120K+ Compounds
120K+ Compounds
 Competitive Price
Competitive Price
 1-2 Day Shipping
1-2 Day Shipping