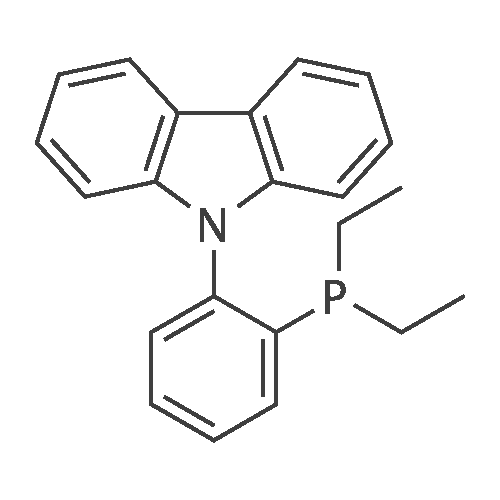
| 29% |
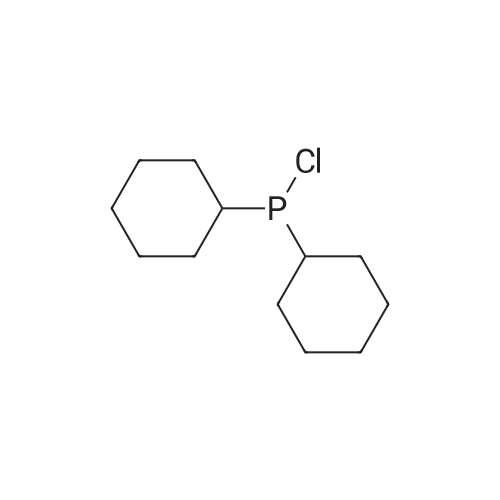
With tri-tert-butyl phosphine; bis(dibenzylideneacetone)-palladium(0); sodium t-butanolate; In toluene; at 100℃; for 20h;Inert atmosphere; Reflux; |
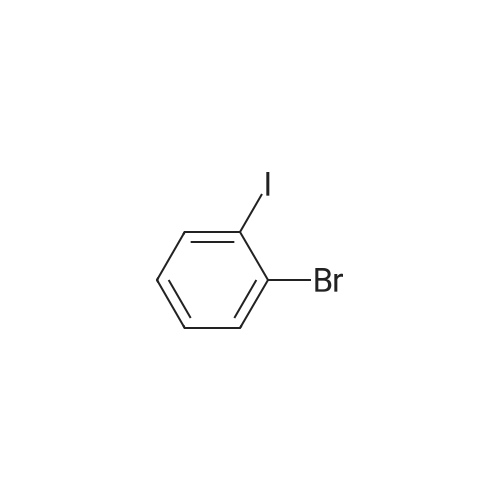
Sigma aldrich in nitrogen (http://www. Sigmaaldrich. Com /) carbazole yarn (50 g, 299. 0 mmol) of toluene 0. 5 L senses a rotation velocity of the disk to, herein 1-bromo-2-iodobenzene sigma aldrich yarn (101. 5 g, 358. 8 mmol), bis (dibenzylideneacetone) palladium (0) (5. 16 g, 8. 97 mmol), tris-tert butylphosphine (3. 02 g, 15. 0 mmol) and sodium tert-butoxide (34. 5 g, 358. 8 mmol) in 100 C sequentially and at the reflux by heating at a 20. Complete after reaction solution at a water doesn't have any error frames, then dichloromethane (DCM) extraction of water to the MgSO4 anhydride after triggers number, filter and was, concentrating it under reduced pressure. Thus-obtained residue number intermediate that is forward separated into flash column chromatography I-3 (27. 9 g, 29%) a obtained. |
| 25% |
With potassium carbonate;copper(l) iodide; In xylene;Heating / reflux; |
Carbazole (1.672 g, 10 mmol), l-bromo-2-iodobenzene (1.5 ml, 12 mmol), potassium carbonate (K CO , 2.7646 g, 20 mmol), copper iodide (CuI, 95 mg, 0.5 mmol), and 25 ml of xylene were refluxed in a nitrogen atmosphere. After cooling to normal temperature was conducted, a product was extracted with ethyl acetate, water was removed with anhydrous magnesium sulfate (MgSO ), and the solvent was removed at a reduced pressure. The resulting product was passed through a silica gel column using a hexane solvent to produce a compound, the solvent was removed at a reduced pressure, and vacuum drying was conducted to produce the resulting white solid compound of Formula a (800 mg, yield 25%).[123] MS: [M+H]+ = 323. |
| 25% |
With potassium carbonate;copper(l) iodide; In xylene;Reflux; Inert atmosphere; |
Preparation Example 1Preparation of Starting Material Represented by Formula aCarbazole (1.672 g, 10 mmol), 1-bromo-2-iodobenzene (1.5 ml, 12 mmol), potassium carbonate (K2CO3, 2.7646 g, 20 mmol), copper iodide (Cud, 95 mg, 0.5 mmol), and 25 ml of xylene were refluxed in a nitrogen atmosphere. After cooling to normal temperature was conducted, a product was extracted with ethyl acetate, water was removed with anhydrous magnesium sulfate (MgSO4), and the solvent was removed at a reduced pressure. The resulting product was passed through a silica gel column using a hexane solvent to produce a compound, the solvent was removed at a reduced pressure, and vacuum drying was conducted to produce the resulting white solid compound of Formula a (800 mg, yield 25%).MS: [M+H]+=323. |
| 25% |
With potassium carbonate;copper(l) iodide; In xylene;Reflux; Inert atmosphere; |
Preparation Example 1Preparation of Starting Material Represented by Formula aCarbazole (1.672 g, 10 mmol), 1-bromo-2-iodobenzene (1.5 ml, 12 mmol), potassium carbonate (K2CO3.2.7646 g, 20 mmol), copper iodide (CuI, 95 mg, 0.5 mmol), and 25 ml of xylene were refluxed in a nitrogen atmosphere. After cooling to normal temperature was conducted, a product was extracted with ethyl acetate, water was removed with anhydrous magnesium sulfate (MgSO4), and the solvent was removed at a reduced pressure. The resulting product was passed through a silica gel column using a hexane solvent to produce a compound, the solvent was removed at a reduced pressure, and vacuum drying was conducted to produce the desired white solid compound (800 mg, yield 25%).MS: [M+H]+=323. |
| 25% |
With copper(l) iodide; potassium carbonate; In N,N-dimethyl acetamide; at 130℃; for 48h;Inert atmosphere; |
General procedure: A mixture of carbazole(1.67 g, 10 mmol), 2-bromoiodobenzene (4.2 g, 15 mmol), K2CO3 (2.76 g, 20 mmol), CuI (0.95 g, 5 mmol) and DMA (100 mL) in a 250 mL three-necked flask was heated at 130 C for 48 h under nitrogen. After cooling, it was poured into water (200 mL) and extracted with CH2Cl2, the combined organic phase was washed with water and dried over MgSO4. After workup, the crude product was isolated by column chromatography (PE) to afford white powder (1.5 g, 25 %); Rf = 0.26 (PE); M. p.: 110 - 112 C; 1H NMR (CDCl3, 500 MHz) delta: 8.17(d, J = 7.7 Hz, 2H), 7.88 (d, J = 8.0 Hz, 1H), 7.52 (m, 2H), 7.43 (m, 3H), 7.31 (t, J = 7.33 Hz, 2H), 7.08 (d, J = 8.1 Hz, 2H); 13C NMR (CDCl3, 125 MHz) delta: 140.84, 136.77, 134.23, 131.13, 130.14, 128.80, 125.92, 123.85, 123.25, 120.33, 119.98, 110.01. |
| 25.5% |
With copper(l) iodide; potassium carbonate; In 5,5-dimethyl-1,3-cyclohexadiene; at 120℃; |
Carbazole (7.39 g, 0.0442 mol), 1-bromo-2-iodobenzene (25 g, 0.0883 mol), CuI (0.421 g, 2.22 mmol), K2CO3 (12.12 g, 0.0883 mol) in a 500 mL 2-neck flask Mol) were dissolved in xylene and refluxed and stirred at 120 C. The mixture was cooled to room temperature, extracted with CH2Cl2, dried over MgSO4, and then the solvent was removed. Hexane solvent to obtain 3.63 g (25.5%) of a white solid. |
| 25% |
With copper(l) iodide; potassium carbonate; In 5,5-dimethyl-1,3-cyclohexadiene;Inert atmosphere; Reflux; |
Under nitrogen atmosphere, a mixture of carbazole (1.7 g), 1-bromo-2-iodobenzene (1.5 ml), potassium carbonate (2.8 g), copper iodide (95 mg), and xylene (25 ml) was refluxed. After cooling to ordinary temperature, the reaction product was extracted with ethyl acetate. The extract was dried over anhydrous magnesium sulfate, and then, the solvent was removed under reduced pressure. The residue was passed through a silica gel column by using a hexane solvent, and then, the solvent was removed under reduced pressure. The obtained residue was vacuum-dried to obtain the intermediate a as a white solid (800 mg, 25% yield). MS: [M+H]+=323 |
| 25% |
With copper(l) iodide; potassium carbonate; In 5,5-dimethyl-1,3-cyclohexadiene;Inert atmosphere; Reflux; |
Under nitrogen atmosphere, a mixture of carbazole (1.7 g), 1-bromo-2-iodobenzene (1.5 ml), potassium carbonate (2.8 g), copper iodide (95 mg), and xylene (25 ml) was refluxed. After cooling to ordinary temperature, the reaction product was extracted with ethyl acetate. The extract was dried over anhydrous magnesium sulfate, and then, the solvent was removed under reduced pressure. The residue was passed through a silica gel column by using a hexane solvent, and then, the solvent was removed under reduced pressure. The obtained residue was vacuum-dried to obtain the intermediate a as a white solid (800 mg, 25% yield). MS: [M+H]+=323 |
| 25% |
With copper(l) iodide; potassium carbonate; In 5,5-dimethyl-1,3-cyclohexadiene;Reflux; Inert atmosphere; |
Carbazole (1.7 g),1-bromo-2-iodobenzene (1.5 ml),Potassium carbonate (2.8 g),Copper iodide (95 mg) and xylene (25 ml) were refluxed under an atmosphere of nitrogen.After cooling to normal temperature, the product was extracted with ethyl acetate.After drying the extract with anhydrous magnesium sulfate,The solvent was removed under reduced pressure.Passed through a silica gel column using a hexane solvent, the solvent was removed under reduced pressure,The obtained residue was dried under vacuum to obtain an intermediate a (800 mg, 25% yield) as a white solid. |
|
With copper(l) iodide; potassium carbonate; In 5,5-dimethyl-1,3-cyclohexadiene;Reflux; Inert atmosphere; |
1.672 g of carbazole, 1.6 mL of 1-bromo-2-iodobenzene, 2.7646 g potassium carbonate, 95 mg of copper iodide, and 25 mL of xylene were refluxed in a nitrogen atmosphere. The mixture was cooled to room temperature, extracted with ethyl acetate, and dried with anhydrous magnesium sulfate to remove moisture, and the solvent was removed under reduced pressure. Silica gel column separation using a hexane solvent was conducted, thus obtaining a compound from which the solvent was then removed under reduced pressure, followed by vacuum drying, yielding 9-(2-bromophenyl)-9H-carbazole as a desired white solid intermediate. |






 Chemistry
Chemistry
 Pharmaceutical Intermediates
Pharmaceutical Intermediates
 Inhibitors/Agonists
Inhibitors/Agonists
 Material Science
Material Science







 For Research Only
For Research Only
 120K+ Compounds
120K+ Compounds
 Competitive Price
Competitive Price
 1-2 Day Shipping
1-2 Day Shipping