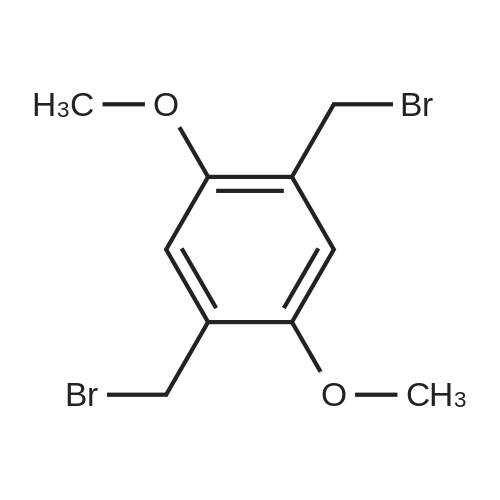
| 45% |
|
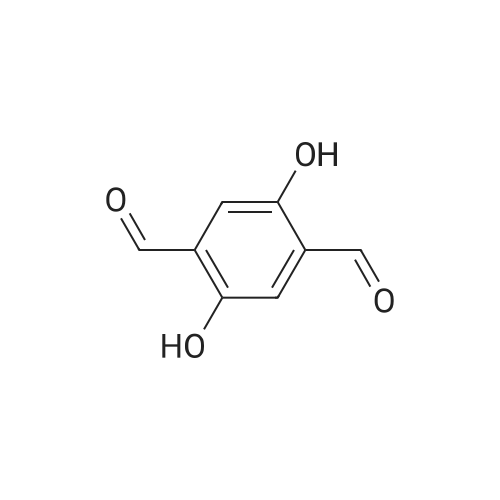
To a solution of 1,4-dimethoxybenzene (1) (10.0 g,72.3 mmol) in 1,4-dioxane (30 mL), HCHO solution (38% inwater, 5 mL) and paraformaldehyde (3.0 g, 99.0 mmol) wereadded in turn [27]. The resulting mixture was heated to90 C, concentrated HCl (2×5 mL) was added during 30 minintervals. Heating continued for 1 h and a further 30 mL ofconcentrated HCl was added. The reaction mixture wascooled to room temperature to afford a white precipitate,which was obtained by filtration and dried under vacuum.The crude product was recrystallized with acetone to giveproduct 1 (4.5 g, 26%) as a white precipitate. A solution ofproduct 1 (15.0 g, 63.8 mmol) and hexamethylenetetramine(18.0 g, 127.6 mmol) in chloroform (50 mL) was stirred atreflux for 24 h. After cooling to r.t., the pale yellow precipitatewas collected by filtration and redissolved in water(30 mL). The aqueous solution was acidified withCH3COOH (10 mL) and stirred at 90 C for 24 h. The mixturewas cooled to r.t. and extracted with DCM (200 mL).The organic phase was washed three times with H2O(200 mL) and dried over anhydrous Na2SO4. After solventevaporation, the residue was recrystallized from CH3CH2OHto yield DMA (5.5 g, 45%) as a bright yellow solid. 1H NMR(300 MHz, CDCl3) delta 10.50 (s, 2H), 7.26 (s, 2H), 3.95 (s, 6H).13C NMR (101 MHz, CDCl3) delta 189.19, 155.73, 129.15,110.92, 77.33, 77.01, 76.70, 56.23. FTMS for C10H10O4:calcd, m/z 195.1 [M+1]+; Anal. Calcd. (194.1). |
| 35% |
|
A mixture of 3 (10.0 g) andhexamethylenetetramine (12.0 g) in chloroform (100 mL) was stirred at 90 C for 24 h. After being cooled to room temperature, the pale yellow precipitate was collected by filtration, washed with CHCl3, dried, and dissolved in water. The aqueous solution was acidified with acetic acid (20 mL) and stirred at 90 C for another 24 h. After cooling, the mixture was extracted with CH2Cl2, and the organic phase was dried over anhydrous MgSO4. After solvent evaporation, the residue was recrystallized from EtOH, giving compound 4 as a yellow solid. Yield: (2.8 g, 35%). 1H NMR (400 MHz, d6-DMSO, 298K,TMS) delta10.4 (s, 2H), 7.44 (s, 2H), 3.94 (s, 6H). 13C NMR (100 MHz, d6-DMSO, 298K, TMS)189.42, 155.65, 129.20, 111.72 ppm. |
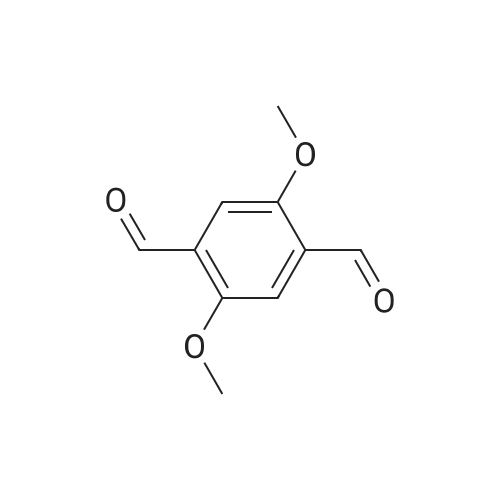
| Ca. 10% |
|
This precursor center was prepared in three synthetic steps, from a procedure involving small modifications of the method already reported [33] . Initially, we dissolved 24.1 g (0.10 mol) of I and 30.0 g (0.21 mol) of urotropine (hexamethylenetetramine) in 200 mL of chloroform. The mixture was refluxed overnight. At the end of this first stage, after leaving the mixture cool to room temperature, the solvent was removed under reduced pressure and the solid residue obtained was dissolved in 320 mL of a 50% acetic acid aqueous solution, being refluxed subsequently for 12 h. In the last part of the process, 25 mL of concentrated HCl was dropwise added to the solution, which was then refluxed for 8 h. After cooling, we observed the formation of a yellow precipitate, which was filtered off, washed with cold water and ethanol and dried under vacuum. Yield: 2.1 g (?10%), m.p. 207 C (literature m.p.: 207 C). From the filtrate, anadditional 0.8 g of the product could be obtained; its m.p. was 200 C. Thesubstance was used in the next step without further purification. Elemental analysis - Percentages found: C, 61.9; H, 5.2. Calcd. for C10H10O4: C, 61.9; H,5.2. Main IR bands (KBr): 3435, 3069, 3048, 2992, 2953, 2932, 2870, 2833, 1679,1503, 1483, 1466, 1408, 1398, 1302, 1131, 1042, 878, 660 cm-1. 1H NMR (CDCl3,300 MHz): 3.93 ppm (s, 6H, -OCH3); 7.44 ppm (s, 2H, aromatic ring); 10.48 ppm(s, 2H, -CHO). |






 Chemistry
Chemistry
 Pharmaceutical Intermediates
Pharmaceutical Intermediates
 Inhibitors/Agonists
Inhibitors/Agonists
 Material Science
Material Science







 For Research Only
For Research Only
 120K+ Compounds
120K+ Compounds
 Competitive Price
Competitive Price
 1-2 Day Shipping
1-2 Day Shipping