| 91% |
With benzonitrile; at 180℃; for 3h; |
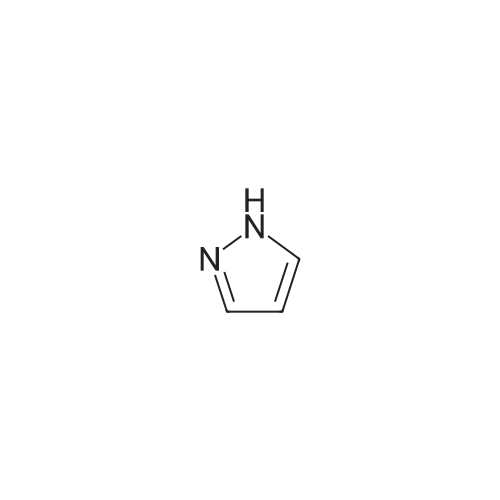
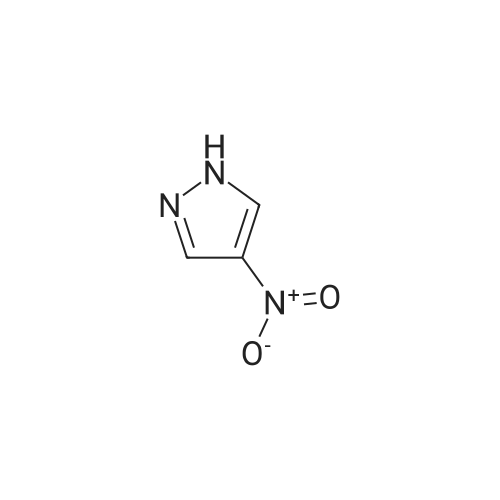
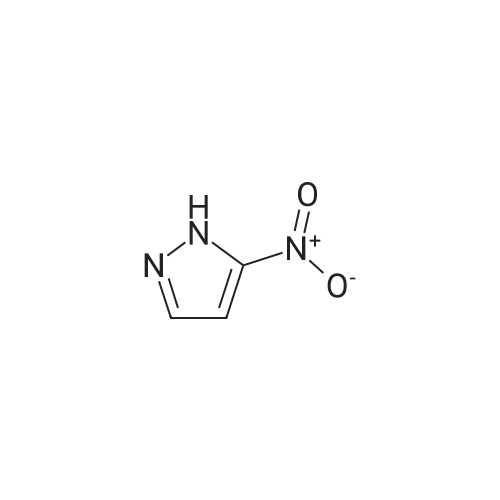
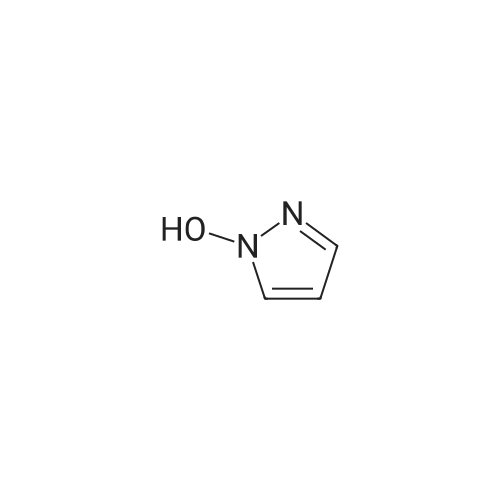
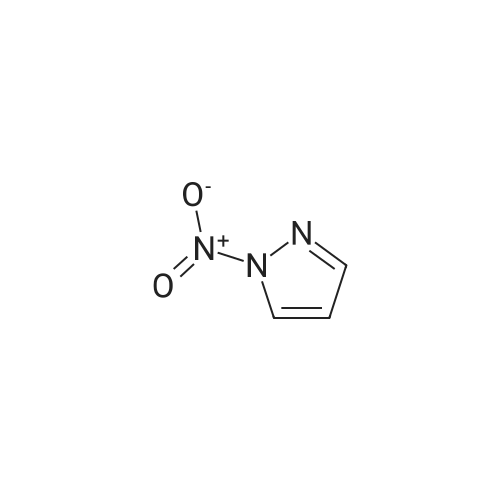
A stirred mixture of <strong>[7119-95-1]<strong>[7119-95-1]1-nitropyrazol</strong>e</strong> (3.45 g, 30.5 mmol) in benzonitrile (33 mL) was heated at 180 °C for 3 h. The mixture was cooled to rt, diluted with hexane and stirred at rt for 20 min. The precipitated solid was collected by filtration to afford 3-nitro-lH-pyrazole as a tan solid (3.16 g, 91percent). H NMR (300 MHz, DMSO- 6) delta 13.94 (br s, 1H), 8.03 (d, J = 2.4 Hz, 1H), 7.03 (t, J = 2.4 Hz, 1H). |
| 79% |
In benzonitrile; for 2h;Reflux; |
Example 5 (S)-3-Cyclopentyl-N-[1-(2-methoxy-2-methyl-propyl)-1H-pyrazol-3-yl]-2-(4-methoxy-2-oxo-2,5-dihydro-pyrrol-1-yl)-propionamide A solution of <strong>[7119-95-1]1-nitro-1H-pyrazole</strong> (4.00 g, 35.4 mmol) in 40 mL of benzonitrile was refluxed for 2 h. After being cooled to 25° C., the mixture was poured into hexanes (160 mL). A white solid precipitated which was filtered and dried in vacuo, to afford 3-nitro-1H-pyrazole (3.16 g, 79percent): H1-NMR (400 MHz, DMSO-d6) delta 7.01 (1H, d, J=2.4 Hz), 8.01 (d, 1H, J=3.4 Hz). |
| 79% |
In benzonitrile; for 2h;Reflux; |
A solution of <strong>[7119-95-1]1-nitro-1H-pyrazole</strong> (4.00 g, 35.4 mmol) in benzonitrile (40 mL) was refluxed for 2 h. After being cooled to 25° C., the mixture was poured into hexanes (160 mL). A white solid precipitated which was filtered and dried in vacuo, to afford 3-nitro-1H-pyrazole (3.16 g, 79percent). 1H-NMR (400 MHz, DMSO-d6) delta 7.01 (1H, d, J=2.4 Hz), 8.01 (d, 1H, J=3.4 Hz). Intermediate 2. |
| 79% |
In benzonitrile; for 2h;Heating / reflux; |
A solution of <strong>[7119-95-1]1-nitro-1H-pyrazole</strong> (4.00 g, 35.4 mmol) in 40 mL of benzonitrile was refluxed for 2 h. After being cooled to 25° C., the mixture was poured into 160 mL of hexanes. A white solid precipitated which was filtered and dried in vacuo, to afford 3-nitro-1H-pyrazole (3.16 g, 79percent). H1-NMR (400 MHz, DMSO-d6) delta: 7.01 (1H, d, J=2.4 Hz), 8.01 (d, 1H, J=3.4 Hz). |
| 79% |
In benzonitrile; for 2h;Heating / reflux; |
Example 15 (E)-4-{3-[2-(3-Chloro-4-methanesulfonyl-phenyl)-2-cyclopentyloxyimino-acetylamino]-pyrazol-1-ylmethyl}-benzoic acid methyl ester A solution of <strong>[7119-95-1]1-nitro-1H-pyrazole</strong> (4.00 g, 35.4 mmol) in 40 mL of benzonitrile was refluxed for 2 h. After being cooled to 25° C., the mixture was poured into hexanes (160 mL). A white solid precipitated which was filtered and dried in vacuo, to afford 3-Nitro-1H-pyrazole (3.16 g, 79percent): H1-NMR (400 MHz, DMSO-d6) delta7.01 (1H, d, J=2.4 Hz), 8.01 (d, 1H, J=3.4 Hz). |
| 56% |
at 145℃; for 10h; |
N-Nitropyrazole (2 g, 18 mmol) was heated at 145 °C for 10 h in a round-bottom flask. Then the product was purified by silica column chromatography using 5:95 v/v methanol/chloroform as eluent to afford 56percent of 3(5)-<strong>[7119-95-1]nitropyrazole</strong> as a white solid. |
| 51% |
In methoxybenzene; at 145℃; |
1-Nitropyrazole (10 g, 88 mmol) was heated in 600 ml of anisole at 145°C overnight. The mixture was cooled in a freezer which caused a precipitate to form. The precipitate was collected to give 5.075 g of the title compound as a tan solid (51percent). [M+H] calc'd for C3H3N3O2, 114; found, 114. |
|
In methoxybenzene; at 145℃; |
3-Nitro-lH-pyrazolew; [539] [Ref.: Janssen, J.W.A.M. and Habraken, C.L., J. Org. Chem., 1971,36,3081.] A solution of l-nitro-ll/'-pyrazole (3.0g, 0.026 mol) in anisole (200mL) was heated at 145°C overnight. The mixture was cooled to rt, the white solid was collected by filtration and washed with hexanes. The mother liquid was diluted with hexanes (500mL) and cooled to -20°C, and the resulting off-white solid was collected and combined with the previous solid.LC-MS (ES, Pos.): 1 14 [MH+]. 'H NMR (DMSO-d6, 400 MHz): 5 = 7.04 (d, J= 2.5 Hz, 1H),8.04 (d, /= 2.5 Hz, 1H), 13.96 (br s, 1H). |
|
In benzonitrile; at 180℃; for 3h; |
A solution of 1.0 g of <strong>[7119-95-1]N-<strong>[7119-95-1]nitropyrazole</strong></strong> in 10 mL of benzonitrile was heated for 3 h at 180 °C, after cooling the reaction mixture was poured into 30 mL of hexane. 3-<strong>[7119-95-1]nitropyrazole</strong> was collected by filtration. The crude yield after washing with hexane and drying was 0.93 g (98percent). The solid was recrystallized from water. m.p. 174?175 °C. FT-IR (KBr, cm?1) 3180 (N?H), 1520 and 1351 cm?1 (NO2). 1H NMR (DMSO-d6) delta: 7.96 (d, 1, 5(3)-H), 6.96 (d, 1, 4-H). EI-MS: m/z 113 (M+·). Anal. Calcd for C3H3N3O2: C, 31.86; H, 2.67; N, 37.16. Found: C, 32.19; H, 2.84; N, 37.18. |
|
at 180℃; |
A 100mL one-neck flask equipped with a magnetic stirrer was added 5g of compound 1 and 15mL acetic acid, stirred, to give a clear solution. Under ice bath 3.5mL of fuming nitric acid was slowly added dropwise to the mixed solution, maintaining vigorous stirring, the reaction temperature does not exceed 30 deg. C. 10mL acetic anhydride was added to the mixture, after the addition, the mixture immediately became cloudy, to give a white turbid liquid. After the addition it was stirred at room temperature, until the solution became clear, about 2.5h. After completion of the reaction, the solution was slowly poured into 100mL crushed ice and with vigorous stirring, potassium carbonate was added portionwise until the mixture was neutral, white precipitate was filtered to give 5.6g of white powdery crystals, the filtrate was extracted with ethyl acetate, the combined organic phase was dried over anhydrous sodium sulfate, the solvent was removed by rotary evaporation to give pale yellow crystals 1.3490g, total crude was 6.9490g, yield 91.9percent recrystallized from benzene. Weigh three 100mg compound 2 into thick-walled pressure bottle, gradually heated to 160 deg. C, 180 deg. C, 200 deg. C, the reaction at this temperature for 1 ~ 3h to obtain compound 3, the results shown in Table 1. |
|
In benzonitrile; at 180℃; for 3.5h; |
<strong>[7119-95-1]N-<strong>[7119-95-1]nitropyrazole</strong></strong> is dissolved in benzonitrile, heated to 180 ° C, incubated for 3.5 h,cooled to 50-60 ° C after completion of thereaction, and added to n-hexane to make 3-nitrate The pyrazole is precipitated, filtered and dried to give 3-<strong>[7119-95-1]nitropyrazole</strong>; |






 Chemistry
Chemistry
 Pharmaceutical Intermediates
Pharmaceutical Intermediates
 Inhibitors/Agonists
Inhibitors/Agonists
 Material Science
Material Science







 For Research Only
For Research Only
 120K+ Compounds
120K+ Compounds
 Competitive Price
Competitive Price
 1-2 Day Shipping
1-2 Day Shipping