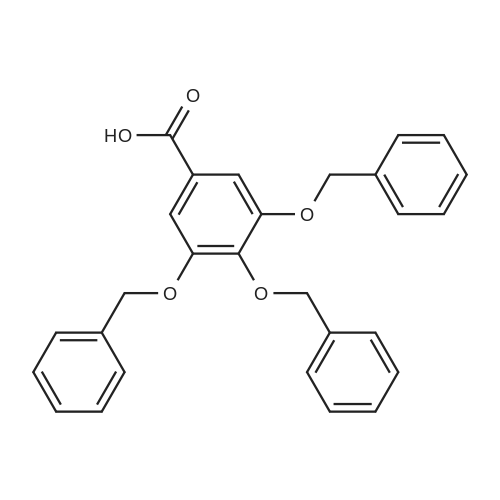
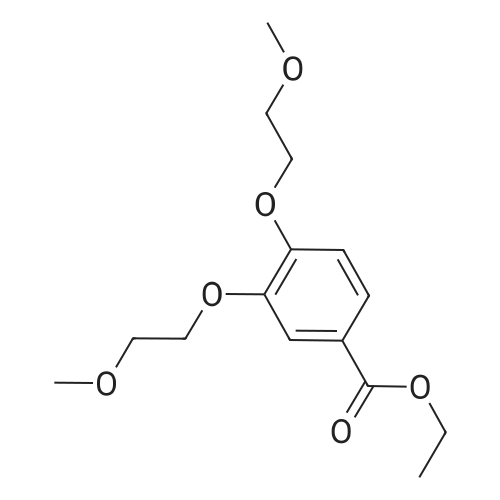
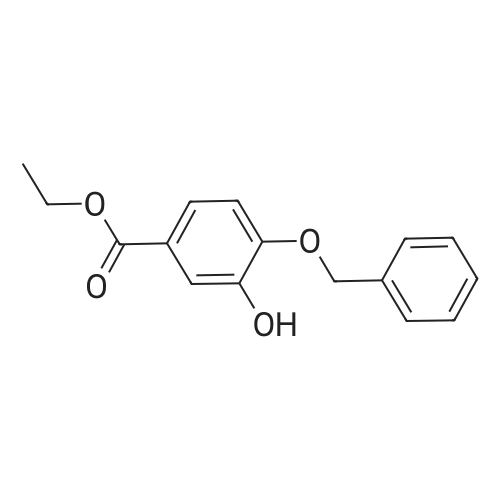
| 95.4% |
With potassium carbonate; In N,N-dimethyl-formamide; at 10 - 60℃; for 4h;Large scale; |
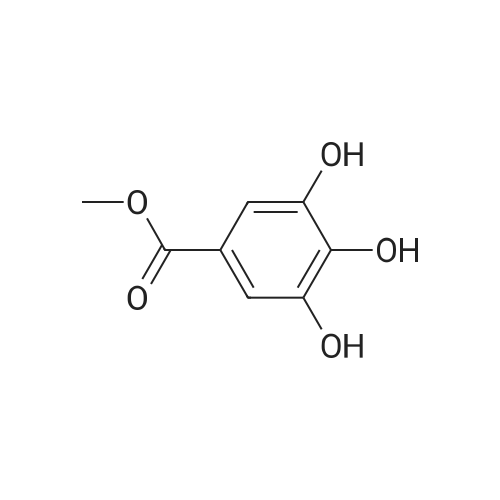
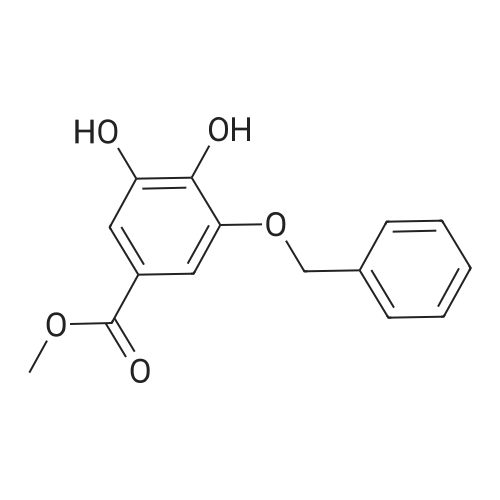
3,4,5-trihydroxybenzoate (9.6 kg, 1.0 eq.) and DMF (76.8 liters) are charged to a reactor at 10-25 C. 2. To the reactor is charged K2C03 (25.1 kg, 3.5 eq.) at the same temperature. 3. Benzyl bromide (28.4 kg, 3.2 eq.) is then added slowly to the mixture at a temperature of from 20-45 C and the mixture aged at about 60 C for about 4 hours. 4. Analysis of the reaction mixture shows that 5. The solids are filtered off and the cake washed with DMF twice (1 vol.). 6. The filtered solution and wash is added to water (115 liters) at 5 C and the mixture stirred for 2 hours at 5-15 C. 7. The resulting mixture was filtered and the cake washed with water. 8. The isolated solid was dried for 12 hours under vacuum at 45 C to obtain the product (22.6 kg as an off-white solid. 9. In this alternate process IIIa-13-1-1 is obtained as an off-white solid with 99.4% purity in a 95.4% yield. |
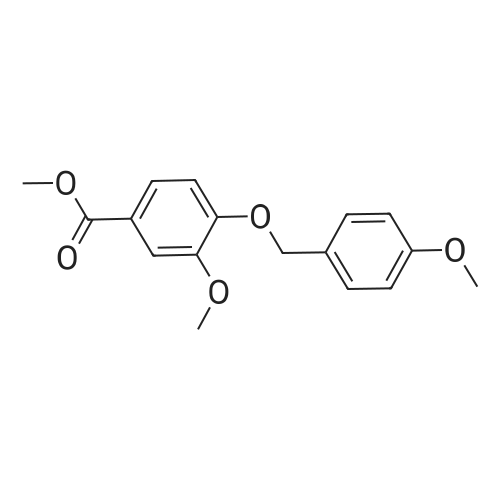
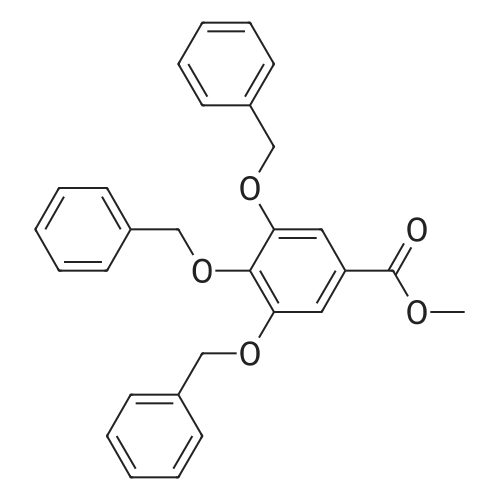
| 94.2% |
With potassium carbonate; In N,N-dimethyl-formamide; at 40℃; |
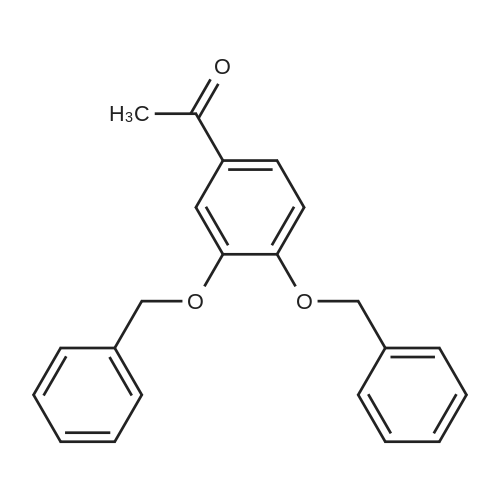
Step A: A solution containing methyl gallate (20.0g, 109mmol), potassium carbonate (90.1g, 652mmol), DMF (120mL), a mixture of benzyl (74.3g, 434mmol) bromide was stirred overnight at 40 ,After the addition of water (240 mL), EtOAc (EtOAc m. The solvent was distilled off under reduced pressure to give 3,4,5 - benzyloxy - benzoic acid methyl ester (68) (46.5g), yield 94.2%. |
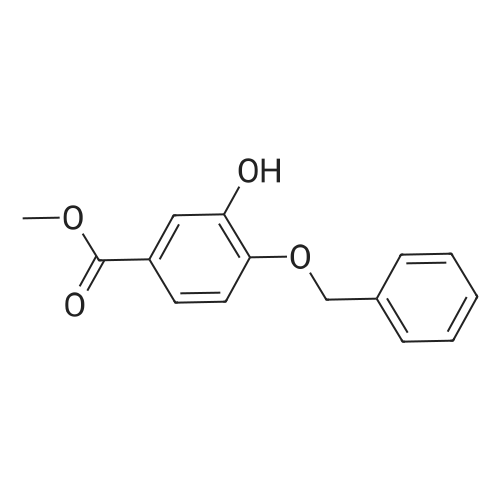
| 90% |
With potassium carbonate; In N,N-dimethyl-formamide; at 60℃; for 15h;Inert atmosphere; |
To a stirred solution of methyl 3,4,5-trihydroxybenzoate 12 (0.94 g, 5.15 mmol) and potassium carbonate (6.56 g, 25.7 mmol)in DMF (20 mL), benzyl bromide (4.4 g, 25.7 mmol) was added. The reaction mixture was stirred at 60 C under N2 atmosphere. After 15 h, water was added, and the whole was extracted with chloroform. The organic layer was washed with brine, dried over sodium sulfate, filtered and concentrated in vacuum to obtain methyl 3,4,5-tri(benzyloxy)benzoate 13 (1.93g, 90%). |
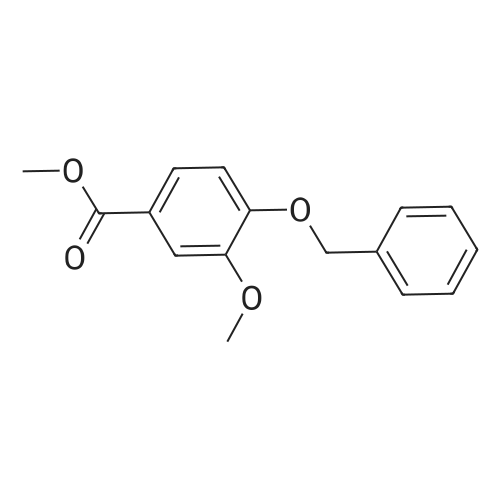
| 89% |
With potassium carbonate; In N,N-dimethyl-formamide; for 6h; |
Compound 2 (1 g, 5.43 mmol) was taken in dry DMF (60 mL) and was stirred in presence of K2CO3 (10 g , 54.34 mmol). Benzyl bromide (4 mL, 16.70 mmol) was added to the stirred solution and stirring was continued for 6h. The progress of reaction was monitored by TLC. After completion of reaction, the reaction mixture was extracted with EtOAc and washed with water. The organic layer was then dried over anhydrous Na2SO4, and the solvent was evaporated to get the crude product, which was purified by column chromatography using using 5% EtOAc in petroleum ether as the eluent to get the pure product 3 as white powder (2.2 g, yield 89 %). 1H NMR (CDCl3, 400 MHz): 7.37-7.35 (m, 4H), 7.33-7.24 (m, 10H), 7.20-7.17 (m, 3H), 5.06 (s, 4H), 5.04 (s, 2H), 3.82 (s, 3H); FTIR (KBr, cm-1 ): 3411, 3044, 3031, 2948, 2878, 1715, 1589, 1499, 1453, 1110 and 754 cm-1 ; mp. 94C |
| 85% |
With potassium carbonate; In N,N-dimethyl-formamide; at 80℃; for 6h; |
General procedure: Gallic acid (1.5 g,8.83 mmol) was dissolved in 9 cm3 methanol, conc.0.45 cm3 sulfuric acid was added thereto, and the mixturewas stirred under reflux for 5 h. The reaction vessel wascooled down to room temperature and the reaction mixturewas neutralized with saturated sodium bicarbonate solutionat 0 C. Then, the organic solvent was removed at reducedpressure, and the residue dissolved in ethyl acetate, washedwith saturated sodium bicarbonate solution, dried overanhydrous sodium sulfate, and concentrated to give methyl1.39 g 3,4,5-trihydroxybenzoate (7.55 mmol, 86% yield)as white solid. This compound was used in the next stepreaction without further purification.Gallic acid (1.5 g,8.83 mmol) was dissolved in 9 cm3 methanol, conc.0.45 cm3 sulfuric acid was added thereto, and the mixturewas stirred under reflux for 5 h. The reaction vessel wascooled down to room temperature and the reaction mixturewas neutralized with saturated sodium bicarbonate solutionat 0 C. Then, the organic solvent was removed at reducedpressure, and the residue dissolved in ethyl acetate, washedwith saturated sodium bicarbonate solution, dried overanhydrous sodium sulfate, and concentrated to give methyl1.39 g 3,4,5-trihydroxybenzoate (7.55 mmol, 86% yield)as white solid. This compound was used in the next stepreaction without further purification.A mixture of methyl 2.114 g 3,4,5-tris(benzyloxy)benzoate(4.66 mmol), sodium hydroxide (96 mmol), 6 cm3MeOH, and 12 cm3 dioxane was heated at reflux for 4 h.The solvent was removed and the resulting residue waspartitioned between water and ethyl acetate. The combinedorganic layers were washed with brine, dried (Na2SO4),and concentrated at reduced pressure to give the titlecompound (1.984 g, 4.51 mmol, 97% yield) as a whitesolid. Spectral data were in accordance with those previouslyreported |
|
With potassium carbonate; In acetone; |
[00102] Scheme 1. Synthesis of the benzyloxybenzoyl chlorides 5 [00103] In terms of the core aromatic ring, pyrogallol 8 (present in 2a) and 3- methoxycatechol 6a (X = OMe, Y = H, present in la) were chosen. Two halogen substituted phenols, 6b (X = H, Y = CI) and 6c (X = F, Y = H) were chosen to provide a p-donor (similar to the methoxy group) but electron withdrawing group. An unsubstituted catechol 6d (X, Y = H) was included. As shown in Scheme 2, each of these phenols was then coupled to each of the acid chlorides 5a-f and several methoxy-substituted benzoyl chlorides. After coupling, the benzyloxy esters were deprotected via catalytic hydrogenation. The overall yields are shown in Table 2. |
|
With tetra-(n-butyl)ammonium iodide; potassium carbonate; In N,N-dimethyl-formamide; at 20℃; for 11h;Inert atmosphere; |
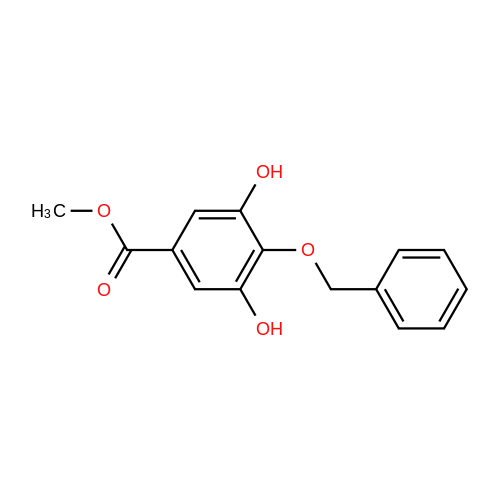
To a solution of 8c (60.0 g, 0.318 mol) in MeOH (638 mL) was added SOCl2 (23.2 mL, 0.319 mol) fordropwise at 60 C. The reaction mixture was stirred at 60 C for 8 h. Then, the reaction mixture wasevaporated under reduced pressure. The crude mixture was applied to following reaction without furtherpurification.To a mixture of crude material (2.00 g, 10.9 mmol) and K2CO3 (6.78 g, 49.0 mmol) in DMF (36.0 mL)were added benzyl bromide (4.10 mL, 35.9 mmol) and TBAI (1.60 g, 4.33 mmol) at room temperature.The reaction mixture was stirred at room temperature for 11 h. Then, the reaction mixture was filteredthrough a pad of Celite, diluted with H2O, and extracted with CH2Cl2. The organic layer was washed withwater and brine, dried over anhydrous MgSO4, and evaporated under reduced pressure. The crude mixturewas applied to following reaction without further purification.To a stirred solution of crude residue in MeOH/THF (1/1, 36.3 mL) was added 4 M NaOH aq. (9.50 mL,38.0 mmol) at 80 C. The reaction mixture was stirred at 80 C for 1 h. Then, the reaction mixture wasquenched with 6 M HCl aq., and filtered. The filtrate was washed with H2O to give 9c (3.30 g, 7.49 mmol,68%) as a white solid.Spectral data for 9c were in good agreement with those reported in reference.16 |






 Chemistry
Chemistry
 Pharmaceutical Intermediates
Pharmaceutical Intermediates
 Inhibitors/Agonists
Inhibitors/Agonists
 Material Science
Material Science







 For Research Only
For Research Only
 120K+ Compounds
120K+ Compounds
 Competitive Price
Competitive Price
 1-2 Day Shipping
1-2 Day Shipping