|
|
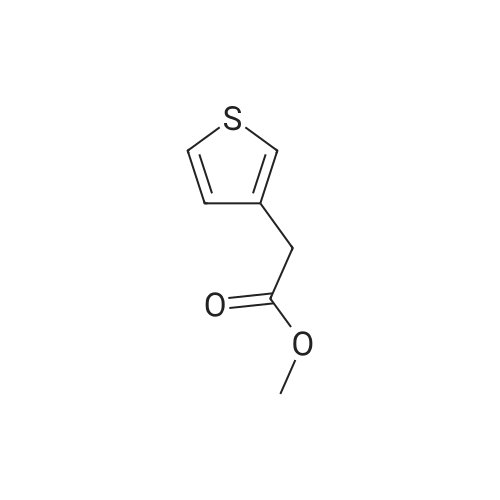
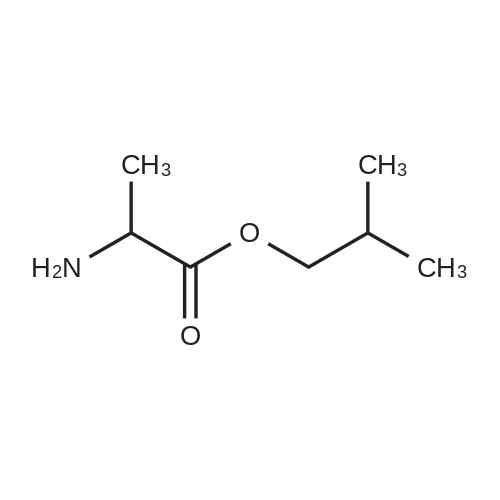
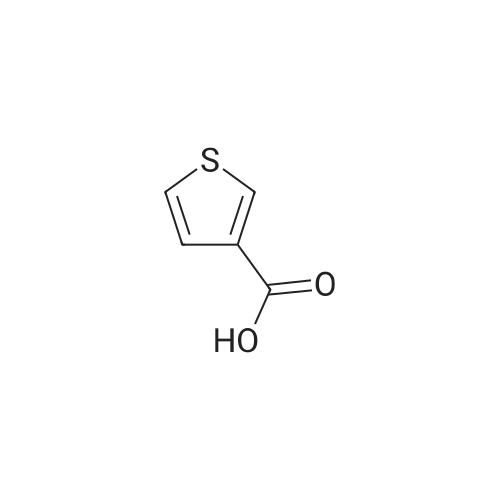
EXAMPLE A60 Synthesis of N-[(3-thienyl)acetyl]alanine iso-butyl ester Following General Procedure I' above, and using <strong>[6964-21-2]3-thiopheneacetic acid</strong> (Aldrich) and alanine iso-butyl ester (prepared following General Procedure J' above), the title compound was prepared. The reaction was monitored by tlc on silica gel and purification was by filtration as described in the general procedure. NMR data was as follows: 1H-nmr (CDCl3): δ=7.33 (m, 1H), 7.14 (m, 1H), 7.01 (m, 1H), 6.09 (m, 1H), 4.58 (m, 1H), 3.88 (m, 2H), 3.60 (s, 2H), 1.91 (m, 1H), 1.37 (d, 3H) 0.92 (d, 6H). |
|
|
EXAMPLE A60 Synthesis of N-[(3-thienyl)acetyl]alanine Iso-butyl Ester Following General Procedure I' above, and using <strong>[6964-21-2]3-thiopheneacetic acid</strong> (Aldrich) and alanine iso-butyl ester (prepared following General Procedure J' above), the title compound was prepared. The reaction was monitored by tlc on silica gel and purification was by filtration as described in the general procedure. NMR data was as follows: 1H-nmr (CDCl3): δ=7.33 (m, 1H), 7.14 (m, 1H), 7.01 (m, 1H), 6.09 (m, 1H), 4.58 (m, 1H), 3.88 (m, 2H), 3.60 (s, 2H), 1.91 (m, 1H), 1.37 (d, 3H) 0.92 (d, 6H). |
|
|
Example A60 Synthesis of N-[(3-thienyl)acetyl]alanine iso-butyl ester Following General Procedure I' above, and using <strong>[6964-21-2]3-thiopheneacetic acid</strong> (Aldrich) and alanine iso-butyl ester (prepared following General Procedure J' above), the title compound was prepared. The reaction was monitored by tlc on silica gel and purification was by filtration as described in the general procedure. NMR data was as follows: 1H-nmr (CDCl3): δ=7.33 (m, 1H), 7.14 (m, 1H), 7.01 (m, 1H), 6.09 (m, 1H), 4.58 (m, 1H), 3.88 (m, 2H), 3.60 (s, 2H), 1.91 (m, 1H), 1.37 (d, 3H) 0.92 (d, 6H). |
|
With 1-(3-(1-pyrrolidinyl)-propyl)-3-ethylcarbodiimide; In chloroform; at 23℃; for 96h; |
Example 60 Synthesis of N-[(3-thienyl)acetyl]alanine iso-butyl ester Following General Procedure I above, and using <strong>[6964-21-2]3-thiopheneacetic acid</strong> (Aldrich) and alanine iso-butyl ester (prepared following General Procedure J above), the title compound was prepared. The reaction was monitored by tlc on silica gel and purification was by filtration as described in the general procedure. NMR data was as follows: 1H-nmr (CDCl3): δ = 7.33 (m, 1H), 7.14 (m, 1H), 7.01 (m, 1H), 6.09 (m, 1H), 4.58 (m, 1H), 3.88 (m, 2H), 3.60 (s, 2H), 1.91 (m, 1H), 1.37 (d, 3H) 0.92 (d, 6H). Optical Rotation: [α]23-52 (c 1 MeOH) ? 589 nm. C13H19NO3S (MW = 269, Mass Spectroscopy (MH+ 269)). GENERAL PROCEDURE IP-EPC coupling P-EPC coupling employs an amino acid ester and a substituted acetic acid compound. The acetic acid derivative is well known in the art and is typically commercially available. The amino acid ester is prepared by conventional methods from the known and typically commercially available N-BOC amino acid as described in GENERAL PROCEDURE J below. Specifically, the appropriate amino ester free base (0.0346 mmols) and substituted phenylacetic acid (0.069 mmols) were dissolved in 2.0 mL CHCl3 (EtOH free), treated with 150 mg of P-EPC (0.87 meq./g) and the reaction was mixed for 4 days at 23C. The reaction was filtered through a plug of cotton, rinsed with 2.0 mL of CHCl3 and the filtrate evaporated under a stream of nitrogen. The purity of each sample was determined by 1H NMR and ranged from 50% to >95%. Between 8.0 and 15.0 mg of final product was obtained from each reaction and was tested without additional purification |






 Chemistry
Chemistry
 Pharmaceutical Intermediates
Pharmaceutical Intermediates
 Inhibitors/Agonists
Inhibitors/Agonists
 Material Science
Material Science







 For Research Only
For Research Only
 120K+ Compounds
120K+ Compounds
 Competitive Price
Competitive Price
 1-2 Day Shipping
1-2 Day Shipping