Abstract: In this study, we developed a metal-free and highly chemoselective method for the reduction of aromatic nitro compounds. This reduction was performed using tetrahydroxydiboron [B2(OH)4] as the reductant and 4,4'-bipyridine as the organocatalyst and could be completed within 5 min at room temperature. Under optimal conditions, nitroarenes with sensitive functional groups, such as vinyl, ethynyl, carbonyl, and halogen, were converted into the corresponding anilines with excellent selectivity while avoiding the undesirable reduction of the sensitive functional groups.
* All experimental methods are cited from the reference, please refer to the original source for details. We do not guarantee the accuracy of the content in the reference.
With tetrafluoroboric acid; sodium nitrite; at 0℃; for 1h;
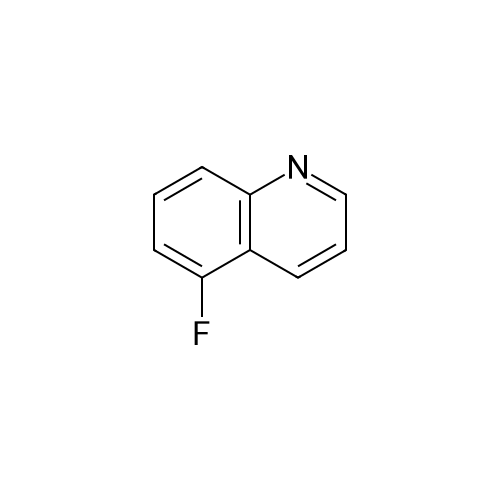
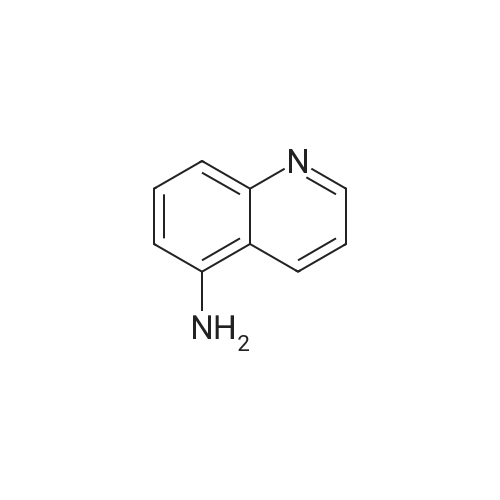
Preparation LIII 5-fluoro-1,2,3,4-tetrahydroquinoline [0363] 5-fluoroquinoline [0364] To a suspension of 5-aminoquinoline (50 g, 347 mmol) in 48percent HBF4 (200 mL) at 0° C. was added portionwise sodium nitrite. This was stirred for 1 hour and then poured into 1:1 ethyl acetate/diethyl ether (500 mL). The resulting suspension was filtered and the solid dried. This solid (82.5 g, 338 mmol) was added portionwise to refluxing xylene (1 L) and stirred for 2 hours then allowed to cool. The xylene was decanted off and the residue dissolved in 1N hydrochloric acid (600 mL). After neutralization with sodium carbonate, the mixture was extracted with ethyl acetate (10.x.500 mL). The extracts were dried over sodium sulfate, filtered and the volatiles removed under reduced pressure. The residue was subjected to silica gel chromatography, eluting with 10-20percent diethyl ether in hexanes. Fractions containing product were combined and concentrated under reduced pressure to provide 28.1 g (55percent) of the desired compound. MS (EI, m/z) C9H6FN (M+1) 148.0 [0365] Reduction [0366] A mixture of 5-fluoroquinoline (28.1 g), 5percent palladium on carbon (5.6 g) in methanol was shaken over night at 40° C. under 60 psi hydrogen. The mixture was filtered through celite and concentrated under reduced pressure. The residue was subjected to silica gel chromatography, eluting with 5-10percent ethyl acetate in hexanes. Fractions containing product were combined and concentrated under reduced pressure to provide 22.5 g (78percent) of the title compound. [0367] MS (EI, m/z) C9H10FN (M+1) 152.0
24.5%
With tetrafluoroboric acid; sodium nitrite; In xylene; at 0℃;Reflux;
To a suspension of 5-aminoquinoline (lO.Og, 0.069 mol) in 48percent HBF4 (40 mL) at 0°C was added portionwise sodium nitrite. This was stirred for 1 hour and then poured into 1 : 1 ethyl acetate/diethyl ether (50 mL). The resulting suspension was filtered and the solid dried. This solid was added portionwise to refluxing xylene (80 mL) and stirred for 2 hours then allowed to cool. The xylene was decanted off and the residue was dissolved in IN aqueous hydrochloric acid (100 mL). After neutralization with sodium carbonate, the mixture was extracted with ethyl acetate (3 x 80 mL). The extracts were dried over sodium sulfate, filtered and the volatiles were removed in vacuo. The residue was purified by silica gel column chromatography, eluting with 2percent ethyl acetate in petroleum ether to afford 5-fluoroquinoline as a colorless oil (2.5 g, 24.5percent).'H-NMR (300 MHz, CDC13) delta 8.93 - 8.98 (m, 1H), 8.43 - 8.46 (m, H), 7.92 (d, / = 8.4 Hz, 1H), 7.62 - 7.78 (m, 1H), 7.41 - 7.49 (m, 1H), 7.22 - 7.26 (m, 1H)
800 mg
With tetrafluoroboric acid; sodium nitrite; at 0℃; for 1h;
To a solution of quinolin-5 -amine (2 g, 13.9 mmol) in 10 mL of 48percent HBF4 at 0°C was added sodium nitrite (933 mg, 13.5 mmol) portionwise. This was stirred for 1 hour and then poured into 1 : 1 ethyl acetate diethyl ether mixture (50 mL). The resulting suspension was filtered and the solid was dried. This solid was added portionwise to refluxing xylene (30 mL) and stirred for 3 hours, then allowed to cool. The xylene was decanted off and the residue was dissolved in IN HC1 (50 mL). After neutralization with NaHC03, the mixture was extracted with ethyl acetate (3 x 50 mL). The extracts were dried over sodium sulfate, filtered and the volatiles were removed under reduced pressure. The residue was purified by silica gel chromatography (3percent EtOAc/PE) to afford 800 mg of title compound as colorless oil. LC-MS: m/z 148.2 (M+H)+
800 mg
With tetrafluoroboric acid; sodium nitrite; at 0℃; for 1h;
To a solution of quinolin-5-amine (2 g, 13.9 mmol) in 10 mL of 48percent HBF4 at 0° C. was added sodium nitrite (933 mg, 13.5 mmol) portionwise. This was stirred for 1 hour and then poured into 1:1 ethyl acetate diethyl ether mixture (50 mL). The resulting suspension was filtered and the solid was dried. This solid was added portionwise to refluxing xylene (30 mL) and stirred for 3 hours, then allowed to cool. The xylene was decanted off and the residue was dissolved in 1N HCl (50 mL). After neutralization with NaHCO3, the mixture was extracted with ethyl acetate (3×50 mL). The extracts were dried over sodium sulfate, filtered and the volatiles were removed under reduced pressure. The residue was purified by silica gel chromatography (3percent EtOAc/PE) to afford 800 mg of title compound as colorless oil. LC-MS: m/z 148.2 (M+H)+
800 mg
With tetrafluoroboric acid; sodium nitrite; at 0℃; for 1h;
To a solution of quinolin-5 -amine (2 g, 13.9 mmol) in 10 mL of 48percent HBF4 at 0°C was added sodium nitrite (933 mg, 13.5 mmol) portionwise. This was stirred for 1 hour and then poured into 1 : 1 ethyl acetate diethyl ether mixture (50 mL). The resulting suspension was filtered and the solid was dried. This solid was added portionwise to refluxing xylene (30 mL) and stirred for 3 hours, then allowed to cool. The xylene was decanted off and the residue was dissolved in IN HC1 (50 mL). After neutralization with NaHC03, the mixture was extracted with ethyl acetate (3 x 50 mL). The extracts were dried over sodium sulfate, filtered and the volatiles were removed under reduced pressure. The residue was purified by silica gel chromatography (3percent EtOAc/PE) to afford 800 mg of title compound as colorless oil. LC-MS: m/z 148.2 (M+H)+
N-(2-chloro-3,4-dimethoxybenzylidene)quinolin-5-amine[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
78%
With acetic acid; In ethanol; at 60℃; for 3h;
General procedure: Substituted aromatic aldehyde (2 mmol) was dissolved in10 mL ethanol and few drop of glacial acetic acid wereadded and reaction mixture was refluxed at 60 C. Then 5-aminoquinoline or 3 -aminoquinoline (1 mmol), were addedwith constant refluxing, and completion of reaction waschecked by TLC analysis. After 3 h, when reaction completed,reaction mixture was poured on to water then a solidappeared. The resultant solid product was filtered andwashed with hexanes. Recrystallization from methanol affordedthe desired solid Schiff bases 1-19
General procedure: In a 1.5 mL reaction vial, B(C6F5)3 (0.025 mmol, 5.0 mol %) was dissolved in chloroform (0.60 mL), towhich diethylsilane (1.75 mmol, 3.5 equiv) was added. After shaking briefly, quinolines (1a-p, 0.50 mmol, 1.0equiv) was subsequently added to the above catalyst solution under argon atmosphere. The reaction mixturewas stirred at 25-65 oC for 6-24 h for the reaction of 1a-h, and at 25-100 oC for 2-24 h for the reaction of 1i-p,then allowed to cool down to room temperature and concentrated under reduced pressure to give the crudeproduct. This reaction mixture was then treated with 0.25 N HCl ethereal solution (7 mL) and stirred at roomtemperature for 1 h to give the solid residue, which was subsequently washed with ether. The solid residue wasthen dissolved or suspended in MeOH (1.0 mL) and neutralized with Na2CO3·H2O (0.5 g) at 0 oC. After stirringfor 2 h, MeOH was removed under reduced pressure, and the neutralized reaction residue was dissolved inCH2Cl2 and washed with brine (5 mL) and water (5 mL). The crude product was then obtained from the organicphase of CH2Cl2 solution and finally purified by column chromatography on silica gel to give 2a-h(EtOAc/Hexane = 1/9) and 2i-p (EtOAc/Hexane = 3/7).
2-chloro-2-fluoro-N-(quinolin-5-yl)acetamide[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
91%
With 2,4,6-tripropyl-1,3,5,2,4,6-trioxatriphosphinane-2,4,6-trioxide; N-ethyl-N,N-diisopropylamine; In dichloromethane; ethyl acetate; at 0 - 20℃; for 1.0h;
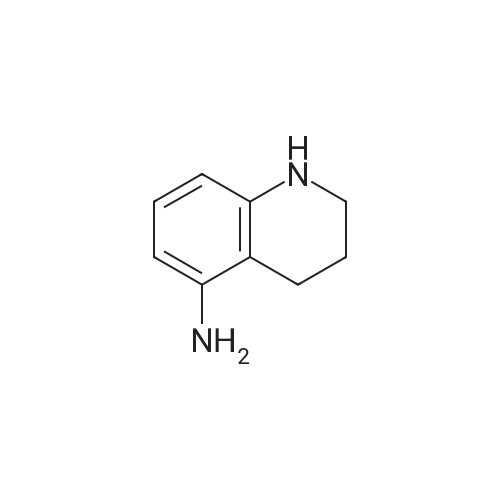
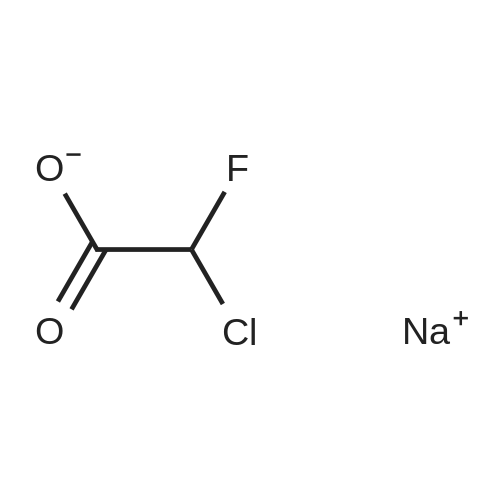
General procedure: To a stirred solution of 5-amino-1-naphthol (126 mg, 0.792 mmol) and <strong>[70395-35-6]sodium chlorofluoroacetate</strong> (159 mg,1.18 mmol) in dichloromethane (8 mL) was added T3P (50 wt%solution in AcOEt, 701 μL, 1.18 mmol) and N,N-diisopropylethylamine(DIPEA) (273 μL, 1.57 mmol) at 0C. Afterstirred at ambient temperature for 1 h, the reaction mixturewas diluted with water and extracted thrice with CHCl3. Thecombined organic layers were washed with brine, dried overNa2SO4, filtered and concentrated in vacuo. The residue waspurified by flash column chromatography on silica gel (hexane/AcOEt = 3 : 1) to afford the title compound (37.2 mg, 18% yield) as a pale purple solid.






 Chemistry
Chemistry
 Pharmaceutical Intermediates
Pharmaceutical Intermediates
 Inhibitors/Agonists
Inhibitors/Agonists
 Material Science
Material Science







 For Research Only
For Research Only
 120K+ Compounds
120K+ Compounds
 Competitive Price
Competitive Price
 1-2 Day Shipping
1-2 Day Shipping