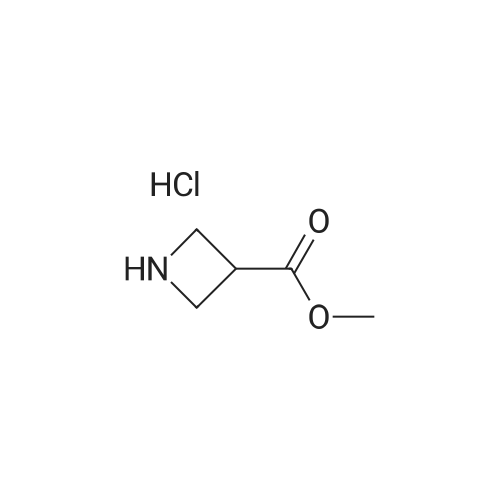
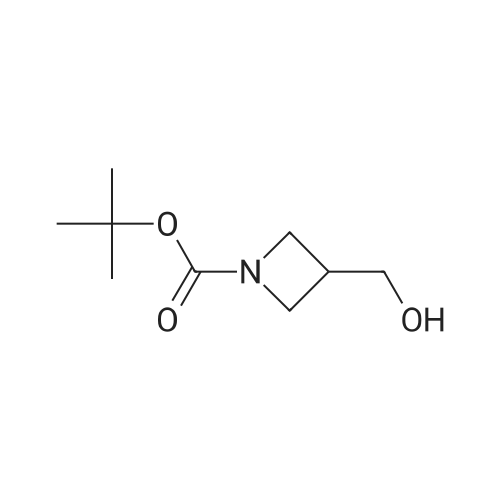
| 95.3% |
|
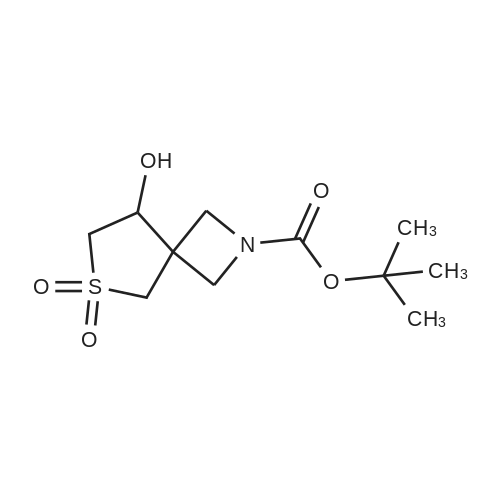
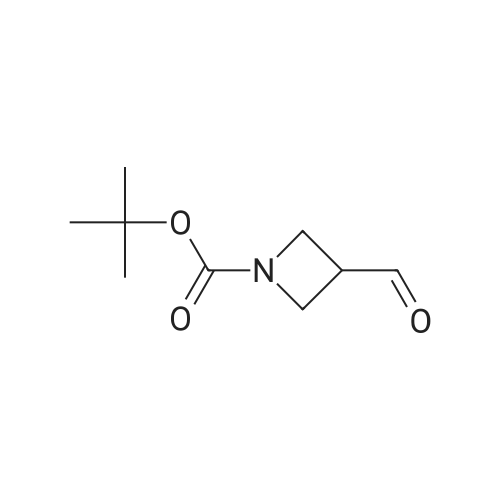
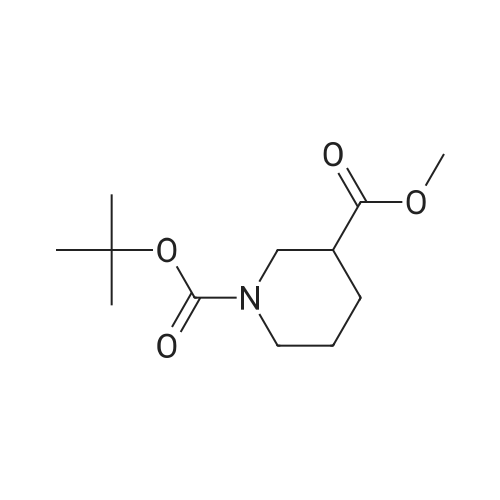
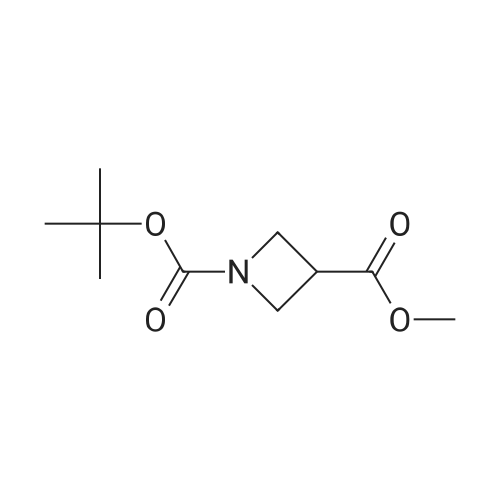
(Production Example 87) tert-Butyl 3-(hydroxymethyl)azetidine-1-carboxylate Lithium aluminum hydride (128 mg) was placed in a round-bottomed flask and suspended in tetrahydrofuran (30 ml). This was cooled in an ice bath, and a solution of <strong>[610791-05-4]methyl 1-tert-butoxycarbonylazetidine-3-carboxylate</strong> (970 mg) in tetrahydrofuran (10 ml) was gradually added thereto, followed by stirring under a nitrogen atmosphere at the same temperature for 1 hr. To the reaction mixture were added water (0.13 ml) and a 5N aqueous solution of sodium hydroxide (0.13 ml) and water (0.39 ml) while cooling in an ice bath, followed by stirring at the same temperature for 1 hr. Insoluble matter in the reaction mixture was removed by filtration. The filtrate was concentrated to provide the titled compound as a colorless oil(805 mg, 95.3 %). 1H-NMR Spectrum (CDCl3) delta (ppm): 1.44 (9H, s), 2.71 (1H, m), 3.69 (2H, dd, J = 5.2, 8.4 Hz), 3.79 (2H, d, J = 6.8 Hz), 4.00 (2H, m). |
| 90% |
With methanol; sodium tetrahydroborate; In tetrahydrofuran; at 80℃; for 1.5h; |
Step 1. tert-Butyl 3-(hydroxymethyl)azetidine-l-carboxylate. Sodium borohydride (756 mg, 20 mmol) was added portionwise into a solution of l-(tert-butyl) 3- methyl azetidine-l,3-dicarboxylate (2.1 1 g, 10 mml) and THF (10 mL). The mixture was heated to 80 C and then MeOH (2 mL) was added very slowly over a 30 minute period. The mixture was stirred for 1 hour, cooled to room temperature and poured slowly into ice-cold HCl (0.5 N). The mixture was extracted (3x) with EtOAc and the organic extracts were dried over anhydrous MgS04. The solvents were removed under vacuum and the residue was purified on silica gel (Biogate; eluting solvents hexanes: EtOAc 2/1 ratio) to afford tert-butyl 3- (hydroxymethyl)azetidine-l-carboxylate as oil (1.69 g, 90% yield):1 NMR (500MHz, CDC13) delta ppm 3.98 (t, J= 8.3, 2H), 3.77 (m, 2H), 3.68 (dd, J= 8.77, 5.37 Hz, 2H), 2.7 (m, |
| 90% |
With sodium hydride; In tetrahydrofuran; at 80℃; for 0.5h; |
General procedure: Triethylamine (0.33 mL, 2.35 mmol) was added into a cold (0 C) mixture of 3-((4'-methoxy- [1,1'-biphenyl]-4-yl)methoxy)azetidine.TFA salt (180 mg, 0.47 mmol), and CH2Cl2 (8 mL). After stirring for 30 minutes cyanogen bromide (99.5 mg, 0.94 mmol) was added and the mixture was allowed to come to room temperature and stirred for 4 h. Then, the mixture was diluted in EtOAc (30 mL) and washed with water and brine. The organics extracts were dried over anhydrous MgSO4. The solvents were removed under vacuum and the residue was purified on silica gel (Biotage; eluting solvents hexanes: EtOAc 3/1 ratio) to afford 3-((4'-methoxy-[1,1'- biphenyl]-4-yl)methoxy)azetidine-1-carbonitrile as white solid (112 mg, 81% yield): |
| 69% |
With lithium aluminium tetrahydride; In tetrahydrofuran; at -15 - 10℃; for 0.5h; |
To a solution of compound 1 (2.5 g) in anhydrous THF (30 mL) was added LiAIH4 (883 mg) at -15 C. The mixture was stirred at -15 C-10 C for 0.5 h, followed by standard work up procedure to give compound 2 (1.5 g, yield 69%) as a colorless oil. |
|
|
<strong>[610791-05-4]1-tert-butyl 3-methyl azetidine-1,3-dicarboxylate</strong> (1055 mg, 4.90 mmol) was dissolved in THF (17 mL) and then cooled to 0 C. MeOH (0.397 mL, 9.80 mmol) and LiBH4 (14.7 mmol) were added sequentially. The reaction was warmed to room temperature over 3 h. Then 10% aqueous potassium sodium tartrate tetrahydrate (Rochelle's Salt) (30 mL) and EtOAc (30 mL) were added and the solution stirred at room temperature over 30 minutes. The organic layer was separated and then dried (Na2SO4) and concentrated to afford 674 mg of t-butyl 3-(hydroxymethyl) azetidine-1-carboxylate (1-3) as a crude product (clear oil). |
|
|
<strong>[610791-05-4]1-tert-butyl 3-methyl azetidine-1,3-dicarboxylate</strong> (1055 mg, 4.90 mmol) was dissolved in THF (17 mL) and then cooled to 0C. MeOH (0.397 mL, 9.80 mmol) and LiBH4 (14.7 mmol) were added sequentially. The reaction was warmed to room temperature over 3 h. Then 10% aqueous potassium sodium tartrate tetrahydrate (Rochelle's Salt) (30 mL) and EtOAc (30 mL) were added and the solution stirred at room temperature over 30 minutes. The organic layer was separated and then dried (Na2SO4) and concentrated to afford 674 mg of f-butyl 3-(hydroxymethyl)azetidine-1-carboxylate (1-3) as a crude product (clear oil). The product was used directly in the next step without purification. |
| 170 kg |
With methanol; sodium tetrahydroborate; In tetrahydrofuran; at 60 - 65℃;Inert atmosphere; Large scale; |
A 5000 liter reactor was charged with sodium borohydride (NaBH4, 72kg, 1902 mol, 1.2 eq.) and tetrahydrofuran (THF, 2240 kg, 6.6 wt), purged with nitrogen gas, and heated to60-65 C. A solution of 3 (377 kg, 90.4 wt%, 1585 mol, 1.0 eq) in methanol (MeOH, 70 kg,2188 mol, 1.40 eq) was added dropwise at 60-65 C with hydrogen gas evolution. Stirring was continued at 60-65 C for 4 to 6 hrs. Gas chromatography sampling indicated all 3 was consumed. More methanol (70 kg, 2188 mol, 1.40 eq) was added dropwise at 60-65 C to quench the excess NaBH4. The reaction mixture was cooled to 3035 C.A second 5000 liter reactor was charged with water (H20, 1700 kg) and heated to 30to 40 C. The reaction mixture in the first reactor containing 3 was transferred to the secondreaction under vacuum to quench the reaction, and stirred at 50C for 1 hrs. Both the organic phase and aqueous phase turned clear. The mixture was cooled to 25-3 0 C and the phases separated. The organic phase was concentrated under vacuum. (60 C, Vacuum: -0.08 Mpa, over about 10 hrs. The aqueous phase was extracted with DCM (1300 kg). The organicphases were combined and washed with 10 w% aq. K2C03 (500 kg x 2) and brine (650 kg),dried with Mg504 (200 kg), and filtered. The filter cake was washed with DCM (260 kg).The wash and filtrate organic phases were combined and concentrated under vacuum to give4 (CAS Reg. No. 142253-56-3, 170 kg, 99.9 % GC purity) as a light yellow oil in 91% yield(uncorrected). The obtained oil became white solid after cooling to room temperature. |






 Chemistry
Chemistry
 Pharmaceutical Intermediates
Pharmaceutical Intermediates
 Inhibitors/Agonists
Inhibitors/Agonists
 Material Science
Material Science







 For Research Only
For Research Only
 120K+ Compounds
120K+ Compounds
 Competitive Price
Competitive Price
 1-2 Day Shipping
1-2 Day Shipping