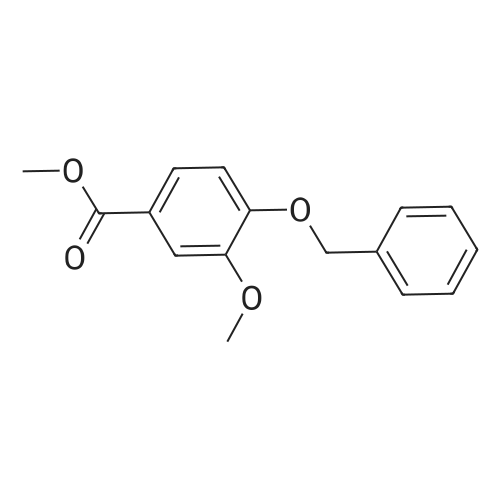
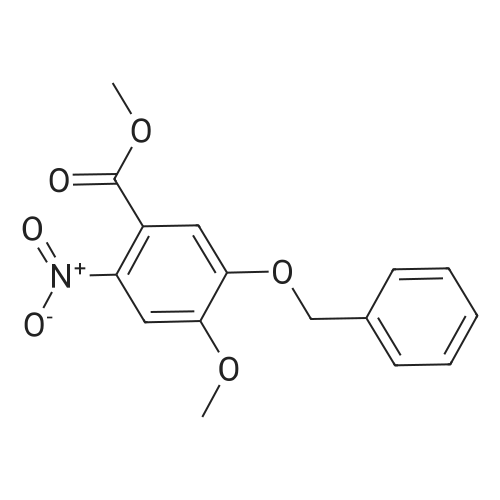
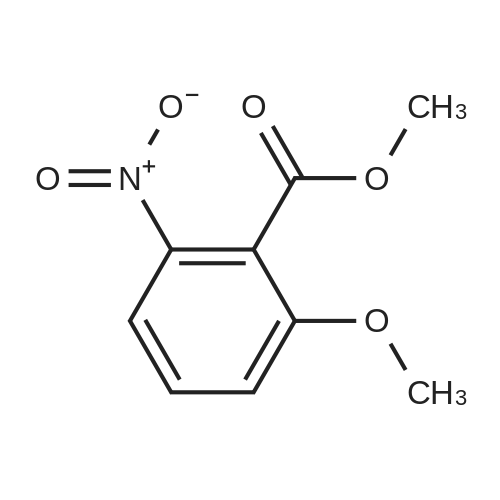
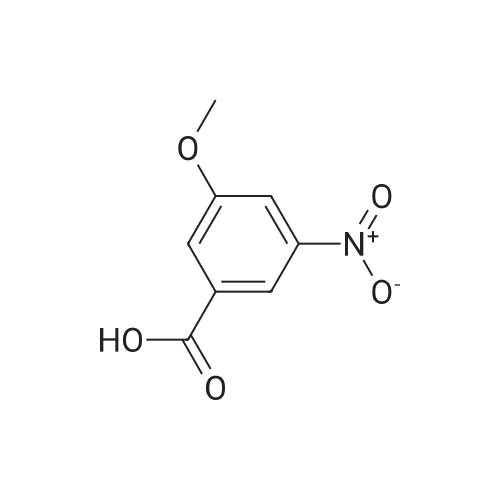
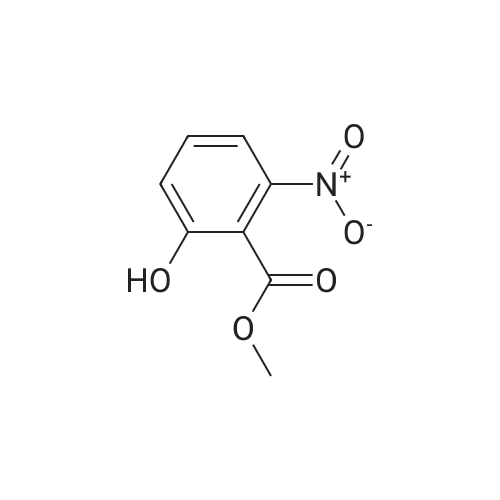
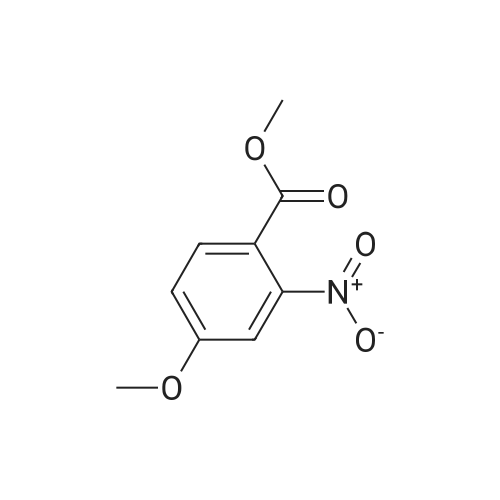
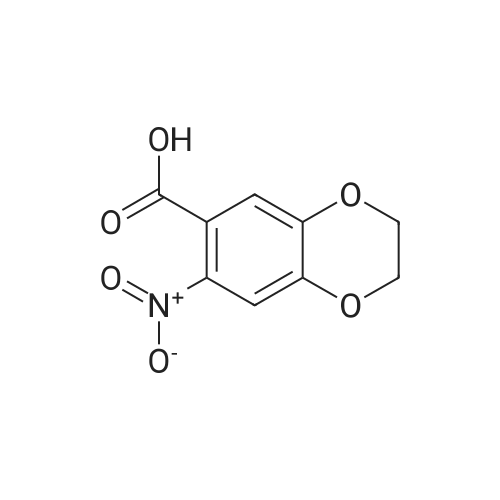
| 97.1% |
With nitric acid; acetic acid; at -10℃; for 0.333333h; |
A mixture of HNO3 (95%, 1000 mL) and acetic acid (1000 mL) was placed in an ice bath and stirred. Compound 119 (440 g, 1.62 mol) in acetic acid (2500 mL) was added dropwise at -10 C. After addition, the mixture was stirred at -10 C. for 20 min, then poured onto a mixture of ice and water (2 L). The mixture was neutralized by the addition of saturated aqueous sodium hydroxide solution to pH 7 and solid precipitated out. The precipitate was collected by filtration, washed with water (300 mL×3) and dried to yield 120 (495 g, yield 97.1%) as a grey solid |
| 95% |
With nitric acid; acetic acid; at 50℃; for 2.0h; |
A 250 mL, round-bottomed flask with a stirring bar, a solution of 4-hydroxy-3-methoxybenzoic acid (20 g, 118.94 mmol) was added slowly to a solution of methanol (100 mL) and concentrated sulfuric acid (10 mL). After being stirred for 12 h at reflux, saturated solution of sodium bicarbonate was added to adjust the pH to 7. Dichloromethane was added and the mixture was then filtered and the organic phase evaporated on a rotary evaporator and to obtain the compound 2 (20.37 g, 94 %). Compound 2 (20.4 g, 111.98 mmol) was added into a 500 mL, round-bottomed flask with a stirring bar, then benzyl bromide (18 mL), potassium carbonate (22 g, 156.8 mmol), DMF (200 mL) were added. It was stirred for 6 h at 80 C. Then the reaction system was poured into right amount of water, white solid (3) was obtained by filtration (28.97 g, 95 %). Compound 3 (16.54 g, 60.74 mmol) was dissolved in CH3COOH (50 mL) and then added into a 250 mL, round bottomed flask with a stirring bar. Then HNO3 (25 mL) was added into the system slowly to keep the temperature of the reaction above 5 C. The reaction temperature was raised to 50 C and kept for another 2 h. After that the system was poured into water and pale yellow solid (I) was obtained6-8 (18.3 g, 95%, m.p.: 134-135 C). |
| 94.7% |
With nitric acid; acetic acid; at 0 - 20℃; for 6.5h; |
Fuming nitric acid (5.8mL, 13.6mmol) was added dropwise to a solution of <strong>[56441-97-5]methyl 3-methoxy-4-benzyloxybenzoate</strong> (4, 3.7g, 13.3mmol) in glacial acetic acid (34mL) at 0-5C and this mixture was stirred at 0-5C for 30min and then for 6hat room temperature. The reaction mixture was poured on ice/ water (120mL) and the resulting precipitate was filtered and washed with chill water, dried to afford 4.0g of the product as a light yellow solid with a yield of 94.7%. Mp: 128-130C; 1H NMR (CDCl3, 300MHz): delta (ppm) 3.90 (s, 3H), 3.95 (s, 3H), 5.23 (s, 2H), 6.89 (s, 1H), 7.28 (s, 1H), 7.33-7.50 (m, 5H). |
| 94.7% |
With nitric acid; acetic acid; at 0 - 20℃; for 6.5h;Cooling with ice; |
34 ml of glacial acetic acid, fuming nitric acid (13.6 mmol, 5.8 ml) was added to a 100 ml round-bottomed flask and stirred at 0 C in an ice bath. Methyl 3-methoxy-4-benzyloxybenzoate 13.3 mmol, 3.7 g) was slowly added to the solution in portions and the reaction was continued at 0 C for 30 minutes and then at room temperature for 6 hours.The reaction solution was slowly poured into 120 ml of ice water, stirred to precipitate solid, filtered, washed with a small amount of ether and dried to give 4.0 g of yellow solid as 2-nitro-4-benzyloxy-5-methoxybenzene acid methyl ester, yield% 94.7 |
| 93.9% |
With nitric acid; acetic acid; at -5 - 25℃; for 16.0h;Cooling with ice; |
will be 53 ml of concentrated nitric acid and 105 ml of acetic acid mixed latter ice-bath lower the temperature to -5 C, will 14.0g compound a is dissolved in 15 ml of acetic acid, then will contain compounds acetic acid solution of concentrated nitric acid and dropping slowly added to the mixed solution of acetic acid, to the reaction solution after the dipping 20-25C, reaction 16 hours, TLC analysis of the, raw materials after complete reaction, the reaction solution is poured into the ice water, extraction with ethyl acetate, washed with saturated sodium chloride solution, the organic phase with saturated sodium bicarbonate solution to alkaline, then saturated sodium chloride solution used for washing, drying by anhydrous magnesium sulphate, filtered filtrate obtained turns on lathe does 15.3g white solid (compound b), this product can be directly used for the next reaction, yield 93.9%; |
| 92% |
With acetic anhydride; copper(II) nitrate; at 0℃; for 6.0h;Inert atmosphere; |
[0509] To a solution of compound Int-1-2 (79.8 g, 0.29 mol) in acetic anhydride (550 mL) under N2 atmosphere at 0C. was added copper (II) nitrate hemi-(pentahydrate) (75.0 g, 0.32 mol). The mixture was stirred at 0C. for 6 h, the reaction was quenched with ice water (800 mL) and a solid precipitated out. The solid was filtered and washed with distilled water (100 mL) and hexane (200 mL2) to obtain compound Int-1-3 (85.5 g, 92%). [0510] 1H NMR (400 MHz, CDCl3) delta 7.52 (s, 1H), 7.45-7.35 (m, 5H), 7.08 (s, 1H), 5.22 (s, 2H), 3.98 (s, 3H), 3.91 (s, 3H). |
| 91% |
|
Step 2. Methyl 4-benzyloxy-3-methoxybenzoate (10.9 g, 40.0 mmol) was converted into methyl 4-benzyloxy-5-methoxy-2-nitrobenzoate (11.6 g, 36.6 mmol, 91%) as described in US 02/0026052 A1, page 51, referent example 15. LC/ESI-MS: m/z=318 [M+H]+; Rt=3.85 min. |
| 91 - 94% |
|
Step 2. Crude material of step 1 (40.0 mmol) was converted into methyl 4-benzyloxy-5-methoxy-2-nitrobenzoate or methyl 5-benzyloxy-4-methoxy-2-nitrobenzoate, respectively, in 91-94% yield as described in US 02/0026052 A1, page 51, reference example 15. |
| 88% |
With nitric acid; tin(IV) chloride; In dichloromethane; at -20℃; for 0.333333h; |
Preparation of 2-nitro-4-benzyloxy-5-methoxy-methylbenzoate 1.15 g of 4-benzyloxy-3-methoxy-methylbenzoate was dissolved in 30 mL anhydrous dichloromethane under a nitrogen atmosphere. The mixture was cooled in a slush bath consisting of carbon tetrachloride and dry ice (-25 C). 6.83 mL of tin(IV)chloride (1.0 M in CH2Cl2) was added to an addition funnel followed by 368 muL of 90% nitric acid. This solution was dripped into the reaction mixture over 5 minutes. The resulting reaction mixture was stirred for 15 minutes in the slush bath, an equal volume of water added and the mixture warmed to room temperature. The resulting layers were separated and the aqueous phase extracted two times with ethyl acetate. The organic phases were combined, dried over sodium sulfate, filtered and concentrated to dryness. The residue was triturated with methanol to give the product as a pale yellow solid in 88% yield. Proton NMR was consistent with the proposed structure. (lit. ref. Organic Letters 1999 vol 1(11) pp 1835-37) |
| 86% |
With nitric acid; In acetic acid; at 50℃; for 5.0h; |
4.1.4 Methyl 4-benzyloxy-5-methoxy-2-nitrobenzoate (12) To a suspension of intermediate 11 (10.1 g, 37 mmol) in AcOH (100 mL) was slowly added 70% HNO3 (20 mL). Subsequently, the mixture was heated to 50 C for 5 h, then cooled to room temperature and poured into ice water (500 mL). After stirring for 1 h, the resulting precipitate was filtered off, washed with water and dried to obtain the nitro compound 12 as a white solid. Yield: 86%, mp: 126-128 C. |
| 70% |
With nitric acid; In acetic acid; for 22.0h; |
To a suspension of <strong>[56441-97-5]methyl 4-(benzyloxy)-3-methoxybenzoate</strong> (141.2 g, 0.519 mol) in acetic acid (400 ml) 160 ml of nitric acid is slowly added. After 20 hours, the reaction is treated with additional nitric acid (100 ml). At 22 hours, the reaction is quenched by addition of ice and then water. The resulting solids are filtered and washed with water three times. Solids are then dried via vacuum oven to provide methyl 4-(benzyloxy)-5- methoxy-2-nitrobenzoate (115 g, 70%). |
| 57% |
With copper(I) nitrate trihydrate; In acetic anhydride; for 2.5h;Cooling with ice; |
Refering to FIG.27, 4-Benzyloxy-5-methoxy-benzoic acid methyl ester 1 (13.6 g, 50 mmol) was dissolved in acetic anhydride (130 mL). Copper nitrate trihydrate (15.1 g, 62.5 mmol) was added in small portions over period of 30 minutes. After stirring for 1 h reaction mixture was poured on ice and stirred for 1 h. Precipitate was filtered off, washed with water and dried thoroughly. Material was recrystallized from ethyl acetate to afford 9.1 g of 2 (57% yield). 1H NMR (500 MHz, CDCl3) : 7.52 (s, 1H), 7.36-7.46 (m, 5H), 7.09 (s, 1H), 5.22 (s, 2H), 3.99 (s, 3H), 3.91 (s, 3H). LC/MS: retention time 3.18 min. (ESI) C16H15NO6 calculated for [M+H]+ 318; found 340 (M+Na). |
| 57% |
With copper(II) nitrate trihydrate; acetic anhydride; for 1.5h; |
4-Benzyloxy-5-methoxy-benzoic acid methyl ester 1 ( 13.6 g, 50 mmol) was dissolved in acetic anhydride (130 mL). Copper nitrate trihydrate (15.1 g, 62.5 mmol) was added in small portions over period of 30 minutes. After stirring for 1 h reaction mixture was poured on ice and stirred for 1 h. Precipitate was filtered off, washed with water and dried thoroughly. Material was recrystallized from ethyl acetate to afford 9.1 g of 2 (57% yield). 1H NMR (500 MHz, CDCl3) delta: 7.52 (s, 1 H), 7.36-7.46 (rn, 5H), 7.09 (s, 1H), 5.22 (s, 2H), 3.99 (s, 3H), 3.91 (s, 3H). LC/MS: retention time 3.18 min. (ESI) C16H16NO6 calculated for [M+H]+318; found 340 (M+Na). |
|
|
Step 2. Crude material of stap 1 (40.0 mmol) was converted into methyl 4-benzyloxy-5-methoxy-2-nitrobenzoate or methyl 5-benzyloxy-4-methoxy-2-nitrobenzoate, respectively, in 91-94% yeld as described in US 02/0026052 A1, page 51, referance example 15. |
|
With nitric acid; In acetic acid; |
REFERENCE EXAMPLE 15 Methyl 5-methoxy-2-nitro-4-(benzyloxy) benzoate Nitric acid (27 mL of a 70% solution) was added dropwise to a suspension of methyl 3-methoxy-4-(benzyloxy) benzoate (14.5 g, 53.0 mmol) in 150 mL of acetic acid. The mixture was stirred at room temperature for 15 minutes and then was heated at 50 C. for 4 hours. The reaction was cooled to room temperature and poured into ice. The precipitate was collected by filtration, washed with water and dried to provide 16.4 g of methyl 5-methoxy-2-nitro-4-(benzyloxy) benzoate as an off-white solid, mp 104-105 C.; MS (ES) m/z 318.1 (M+1). Analysis for C16H15NO6: Calcd: C, 60.57;H, 4.76; N, 4.41. Found: C, 60.39;H, 4.70; N, 4.28. |
|
With nitric acid; In acetic acid; |
Commercially available methyl 4-hydroxy-3-methoxy-benzoate 1 was converted in 5 steps to 4-(3-fluoroanilino)-6-methoxy-7-benzyloxy-quinazolme 15 (35% overall yield).[17,18] The only significant modification involved an improved workup of the nickel chloride hexahydrate-sodium borohydride reduction of the nitro intermediate. Debenzylation with trifluoroacetic acid to give the 7-hydroxy intermediate 16, followed by alkylation with propargyl bromide in acetonitrile, gave 13 in a 65% yield for 2 steps. This method involved an additional step, and the overall yield was greater. In addition, the final 7-hydroxy intermediate 16 could be used to generate other products [19]. |
|
With nitric acid; In acetic acid; at 0 - 60℃; for 1.16667h; |
To a solution of <strong>[56441-97-5]4-benzyloxy-3-methoxy-benzoic acid methyl ester</strong> (7.08 g, 26 mmol) in acetic acid (50 ml), at O0C, was added dropwise nitric acid (7 ml) over 10 minutes. The reaction mixture was warmed to room temperature and then stirred at 60 0C for 1 hour. The reaction mixture was poured into water, extracted with DCM (3x125 ml) and the combined organic extracts were washed with water, saturated aqueous NaHCO3, brine, dried (MgSO4) and concentrated to provide the title compound as a pale yellow solid (7.5 g). LC/MS purity: 95 %, m/z 318 [M+H]+. 1H NMR (300 MHz, CDCI3) delta: 7.55 (1 H, s), 7.53-7.39 (5H, m), 7.11 (1H, s), 5.23 (2H, s), 3.99 (3H1 s), 3.92 (3H, s). |






 Chemistry
Chemistry
 Pharmaceutical Intermediates
Pharmaceutical Intermediates
 Inhibitors/Agonists
Inhibitors/Agonists
 Material Science
Material Science







 For Research Only
For Research Only
 120K+ Compounds
120K+ Compounds
 Competitive Price
Competitive Price
 1-2 Day Shipping
1-2 Day Shipping