| 91.5% |
In toluene; at 90℃; for 5h; |
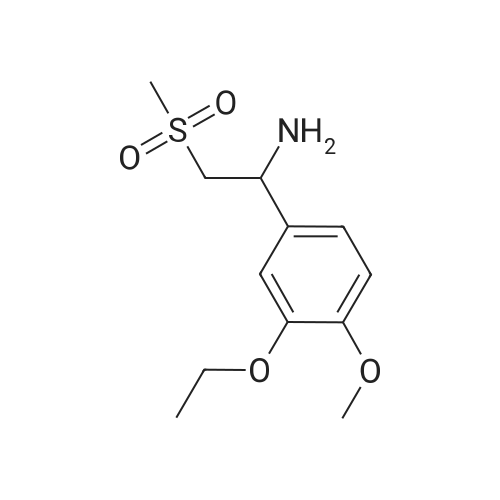
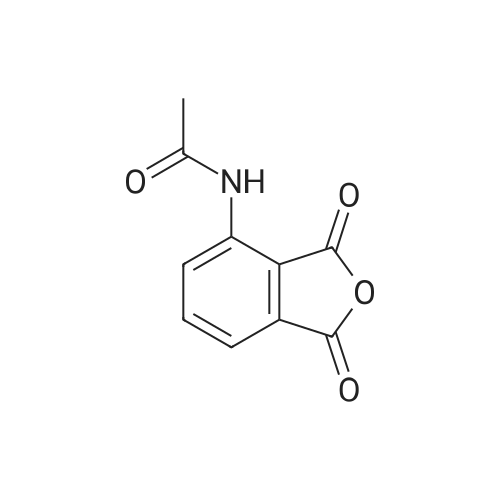
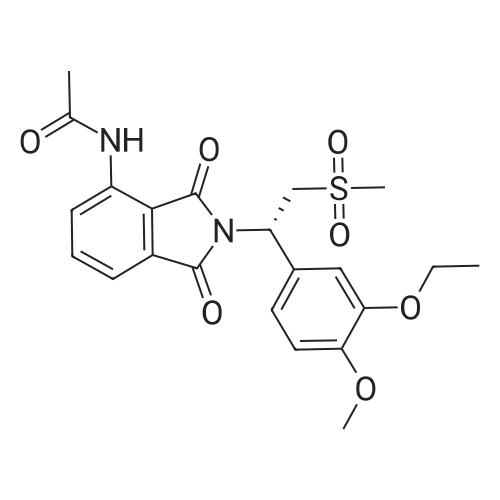
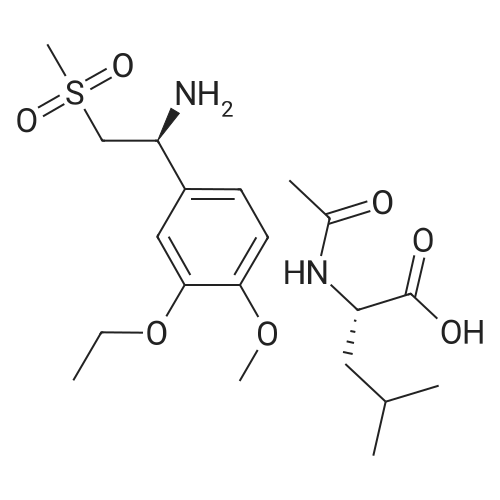
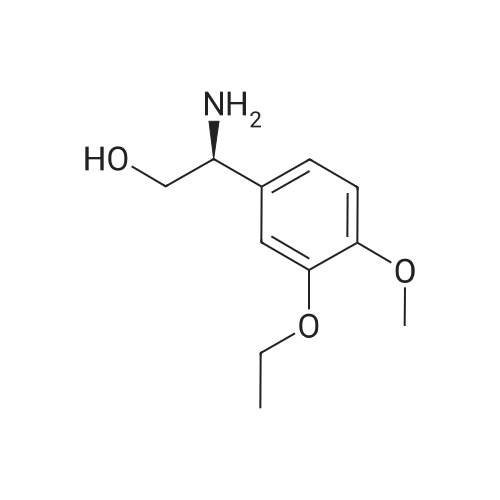
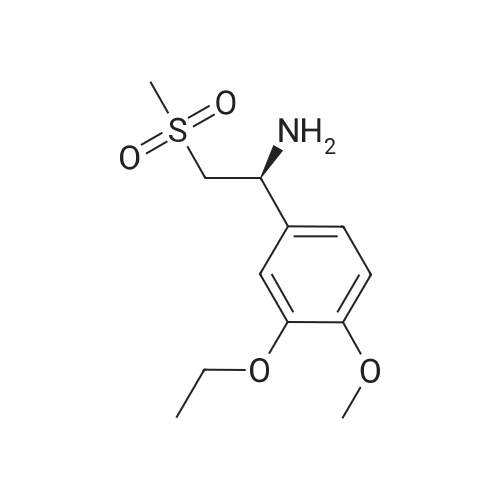
148.7 g of N-acetyl-L-leucinate (0.4% isomer content) of chiral amine intermediate II, 71.38 g of anhydride intermediate III, 1487 ml of toluene were charged into the reaction flask;Heated to 90 ± 5 , incubated for 5 hours, concentrated under reduced pressure to remove toluene; added to 3000ml of ethanol, heating; steamed out of ethanol until evaporated to dryness;Add 1000mL of ethanol, 500mL acetone, heated to reflux, dissolved until clear, add 1.5g activated carbon decolorization, slowly cooled to 15 ; incubated at 10 ± 5 for 6 hours;Filter and wash the filter cake with 200 mL of ethanol. Drying in vacuo at 50 ± 5 C for 4 hours afforded 139.1 g of aphthous (I) in 91.5% yield.After testing, Purcert purity of 99.94%, 0.05% isomer content, crystal form B (see Figure 1). |
| 89% |
With perchloric acid; acetic acid; at 85℃; for 2.5h;Reflux; |
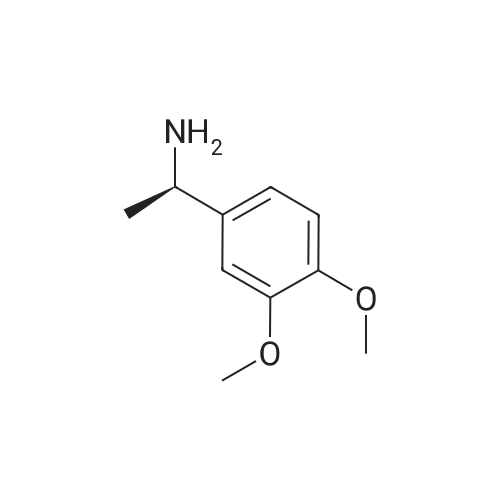
Example 8: Preparation of S-apremilast (I) In a dry 250mL glass flask was added 2.5g of 3-acetamidophthalicanhydride, 25g of glacial acetic acid, 0.2g of perchloric acid, and 2.7g of (S)-1-(4-methoxy-3-ethoxyphenyl)-2-methanesulfonylethylamine (II) (prepared in Example 7). The reaction was heated at 80-85C for 30 minutes then heated at reflux for 2 hours. Glacial acetic acid was recovered through vacuum distillation. The remaining material was then cooled to room temperature 20C. 10g of saturated brine and 20g of ethyl acetate were added and allowed to seperate under stirring. The organic phase was separated. The aqueous phase was extracted once with 20 g of ethyl acetate. The combined organic phases, recovered by distillation of ethyl acetate, gave 4.1g of solid S-apremilast (I). Yield 89.0%, e.e% to 99.8%. |
| 89% |
With acetic acid; for 3h;Reflux; |
3.17 g (11,6 mmol; 99.4 % ee) of (S)-1-(3-ethoxy-4-methoxyphenyl)-2- (methylsulfonyl)-ethylamine of formula (S)-2a (see Example 9), 2.50 g (12.2 mmol) of 3- acetamidopbtalic anhydride of formula S and 52 ml of glacial acetic acid were placed into a250 ml flask. The mixture was refluxed for 3 h and cooled down to 20C. After that, 4 x 25 ml of water were gradually added, under continuous stirring the mixture was inoculated with crystals of Apremilast of formula 1 and stirred at 20 C. The separated crystals were aspirated, washed with a mixture of acetic acid - water (volume ratio 2:5) and dried at a reduced pressure. The amount of 4.75 g of yellowish crystals of the product of formula 1 was obtained(yield 89%, >99% ee, HPLC 99.1%). |
| 84% |
With acetic acid; at 120℃; |
1 Anhydride (4.16 g, 20.26 mmol), chiral amine (19.3 mmol) were added to a 250 mL reaction flask, glacial acetic acid (65 mL) was added, refluxed at 120 C., and thin layer chromatography (TLC) detection reaction ( Methanol: Dichloromethane volume fraction ratio = 1:10). The chiral amine material was completely reacted (about 5-6 h). Acetic anhydride (60 mmol) was added. The reaction was continued for 1 h. The glacial acetic acid was distilled off, and the residue was poured into 100 mL. Intensely stirred water forms a suspension. After suction filtration, the filter cake was washed with 4×50 mL of water, and the filter cake was dried in a drying oven at 50 C. for approximately 4-5 h to obtain 8.0 g of crude yellow appress. The purity was 98.8 by HPLC. %, the deacetylated by-product content is about 0.05%.The above crude product was added to ethanol (70 mL), heated to reflux for 2 h, and then slowly lowered to room temperature with stirring. The solid precipitated and was filtered under suction. The filter cake was washed with ethanol (2×10 mL) and dried (60 C., dried for 3 h). ,getApstThe product is 7.5g, and the total yield is 84%. |
| 81.5% |
With acetic acid; for 6h;Reflux; |
The reaction flask was charged with 350 mL of glacial acetic acid,(0.22 mol) of (1S) -1- (3-ethoxy-4-methoxybenzene) Yl) -2- (methylsulfonyl) ethylamine (formula V)57.4 g (0.28 mol) of 3-acetylaminophthalic anhydride,Stir well, reflux reaction for 6 hours,The reaction was stopped and the residue after dissolution was dissolved in 400 ml of ethyl acetate,Washed with saturated aqueous sodium carbonate (300 mL x 2 times)Washed with brine (150 mL x 1 time).After further desolvation, a mixed solvent of 400 mL of absolute ethanol and 150 mL of acetone was added and stirred for 5 hours,Filter, filter cake with 100mL anhydrous ethanol washing, drying was 82.6g products,The molar yield was 81.5%, the HPLC purity was 98.3%Ee value of 97.9%. |
| 73.1% |
With acetic acid; at 115 - 125℃; |
(1S)-i -(3 -ethoxy-4-methoxy-phenyl)-2-methanesulfonyl- ethyl amine (Formula-V) (125gm) and N-(1, 3-dioxo-1, 3dihyro-2-benzofuran-4-yl) acetamide (Formula-IV) (98.5gm) in to methyl isobutyl ketone (lSOOml) and acetic acid (SSOml) mixture of solvent at ambient temperature. Raise temperature up to 115-120C (115-125C). Remove the water formed during the reaction. After completion of reactiondistill out reaction mass completely. Charge ethyl acetate for extraction and wash with sodium bicarbonate solution and sodium chloride solution. Distill out reaction mass completely and charge acetone followed by ethanol. Raise temperature up to reflux and maintain 1 hr. Reaction gradually cool to ambient temperature and maintain reaction mass and stir for 7-8 hr. Filter the product and wash with ethanol. Charge wet cake into acetone followed by ethanol. Raise temperature up to reflux and maintain 1 hr. Reaction gradually cool to ambient temperature and maintain reaction mass and stir for 7-8 hr. Filter the product and wash with ethanol resulting Formula I with 73.1%yield and 99.98% HPLC purity and 99.99 Chiral purity. The titled product having XRPD values as, 10.1, 12.4, 13.5, 20.8, 22.5, 24.7, and 27.0 ±0.2 (Form-B). |
| 64% |
With acetic acid;Reflux; |
Example 8: Preparation of (S)-1-(3-ethoxy-4-methoxyphenyl)-2-methylsulfonylethyl)-4-amino- isoindoline-1 ,3-dione (apremilast) from the compound of formula VII Into a reaction vessel equipped with magnetic stirrer and condenser was placed (S)-1 -(3- ethoxy-4-methoxyphenyl)-2-(methylsulfonyl)ethanamine (2.24 mmol, 1 g) and was dissolved in 10 mL of glacial acetic acid. Afterwards 3-acetamidophthalic anhydride was added (2.35 mmol, 0.48 g) and reaction mixture was vigorously stirred at reflux overnight. Reaction system was cooled down to room temperature and acetic acid was evaporated under reduced pressure. The organic residue was extracted with dichloromethane, organic phases were washed with water, dried over Na2S04 and solvent was evaporated. The residue was recrystallized from acetone/ethanol mixture, solid material was filtered off and dried in vacuo at 60 C affording 0.66 g (64% Yield) of final product with > 99% ee. 1 H NMR analysis was in agreement with known data. |
| 64% |
With acetic acid;Reflux; |
Example 11 : Preparation of (S)-1 -(3-ethoxy-4-methoxyphenyl)-2-methylsulfonylethyl)-4-amino- isoindoline-1 ,3-dione (apremilast) from the compound of formula 5S Into a reaction vessel equipped with magnetic stirrer and condenser was placed (S)-1 -(3- ethoxy-4-methoxyphenyl)-2-(methylsulfonyl)ethanamine (2.24 mmol, 1 g) and was dissolved in 10 mL of glacial acetic acid. Afterwards 3-acetamidophthalic anhydride was added (2.35 mmol, 0.48 g) and reaction mixture was vigorously stirred at reflux overnight. Reaction system was cooled down to room temperature and acetic acid was evaporated under reduced pressure. The organic residue was extracted with dichloromethane, organic phases were washed with water, dried over Na2S04 and solvent was evaporated. The residue was recrystallized from acetone/ethanol mixture, solid material was filtered off and dried in vacuo at 60 C affording 0.66 g (64% Yield) of final product with > 99% ee. 1 H NMR analysis was in agreement with known data. |
| 59% |
With acetic acid; for 15h;Reflux; |
A stirred solution of 1-(3-ethoxy-4-methoxyphenyl)-2-methylsulfonylethylamine (1.0 g, 3.7 mmol) and 3-acetamidophthalic anhydride (751 mg, 3.66 mmol) in acetic acid (20 mL) was heated at reflux for 15 h. The solvent was removed in vacuo to yield an oil. Chromatography of the resulting oil yielded the product as a yellow solid (1.0 g, 59% yield): mp, 144 C; 1H NMR (CDCl3) delta: 1.47 (t, J=7.0 Hz, 3H, CH3), 2.26 (s, 3H, CH3), 2.88 (s, 3H, CH3), 3.75 (dd, J=4.4, 14.3 Hz, 1H, CH), 3.85 (s, 3H, CH3), 4.11 (q, J=7 Hz, 2H, CH2), 5.87 (dd, J=4.3, 10.5 Hz, 1H, NCH), 6.82-6.86 (m, 1H, Ar), 7.09-7.11 (m, 2H, Ar), 7.47 (d, J= 7 Hz, 1H, Ar), 7.64 (t, J= 8 Hz, 1H, Ar), 8.74 (d, J= 8 Hz, 1H, Ar), 9.49 (br s, 1H, NH); 13C NMR (CDCl3) delta: 14.61, 24.85, 41.54, 48.44, 54.34, 55.85, 64.43, 111.37, 112.34, 115.04, 118.11, 120.21, 124.85, 129.17, 130.96, 136.01, 137.52, 148.54, 149.65, 167.38, 169.09, 169.40; Anal Calc'd. for C22H24NO7S: C, 57.38; H, 5.25; N, 6.08. Found: C, 57.31; H, 5.34; N, 5.83. |
|
With acetic acid;Reflux; |
N-(1,3-dioxo-1,3-dihydroisobenzofuran-4-yl)acetamide, which may be obtained via techniques known in the art, (S)-1-(3-ethoxy-4-methoxyphenyl)-2-(methylsulfonyl)ethanamine, and glacial acetic acid is refluxed overnight and then cooled to <50 C. The solvent is then removed in vacuo, and the residue is dissolved in ethyl acetate. The resulting solution is washed with water, saturated aqueous NaHCO3, brine, and dried over sodium sulphate. The solvent is evaporated in vacuo, and the residue is recrystallized from a binary solvent containing ethanol and acetone. The solid is isolated by vacuum filtration and washed with ethanol. The product is then dried to afford H-CH2-CH3-Compound A. |
| 8.5 g |
With acetic acid; In toluene; at 100 - 105℃; |
(S)- 1 -(3-ethoxy-4-methoxyphenyl)-2-(methylsulfonyl)ethylamine (6.5 g, 0.023 moles) were added to toluene (120 ml) and stirred at room temperature. Acetic acid (30 ml) and N-acetylphthalic anhydride (5.12 g, 0.024 moles) was added to the reaction mixture at room temperature. The temperature of reaction mixture was raised to 100-105C and maintained for 8-10 hours. Distilled the solvent at below 60C under vacuum and cooled the reaction mixture to room temperature. Methyl ethyl ketone (150 ml) was added to the reaction mixture at room temperature. The temperature of reaction mixture was raised to 50-60C and 10% sodium bicarbonate solution (150 ml) was added followed by water (50 ml). The layers were separated and the organic layer was distilled upto one-fourth of its initial volume under vacuum and cooled to 0-5C. The reaction mixture was maintained at 0-5C for 1-2 hours. The solid was filtered, washed with methyl ethyl ketone (15 ml) and dried in oven to provide apremilast as product.Yield: 8.5 g |
|
|
A reaction vessel was charged with glacial acetic acid (300 ml_), Tri ethyl amine (40.72 g), (S) - 1 -(3-Ethoxy-4-methoxyphenyl)-2-methylsulfonylethylamine (100 g) and 3-Acetamido phthalic anhydride (75.05g). Reaction mass was heated for 1.0 hour at 90±3C. After completion of the reaction; reaction mass was cooled to 27 ± 3C. Process water (500 ml_) and dichloromethane (300 ml_) were added in to the reaction mass. Reaction mass was stirred and settled. Layers were separated. Acetyl chloride (17.23 g) was charged in to the organic layer and was heated for 15-20 min at 40 ± 3C. The organic layer was cooled to 27 ± 3C and washed with sodium bicarbonate solution in water. Benzyl alcohol (1 18.68 g) was charged into the organic layer and solvent was distilled out completely under reduced pressure at 42 ± 3C. Methanol (200 ml_) was charged into reaction mass at below 40C. Reaction mass was stirred for 15-20 min. at 52 ± 3C and 1 hour at 27 ± 3C. The solid was filtered and washed with methanol. |
|
With acetic acid; at 85℃; for 5h; |
In a three-neck flask, 500 g of chiral amine and 412 g of anhydride were added.Glacial acetic acid 4L, heated at 85 C under reflux for 5 h.The system was cooled to room temperature and extracted and washed.The solvent was evaporated to dryness to obtain 750 g of crude Apstar.Purity (HPLC) 96%;_(2)Crude refiningThe crude product (200 g) was added to 2-butanone (3L) and water (1L).Heat to 50-60C to dissolve the solids.Cool to 0 ~ 10 C crystallized 1h, suction filtration,175g of refined white crystals are dried at 50C.Yield 87.5%, purity (HPLC) 99.8%,The largest single miscellaneous 0.02%. |






 Chemistry
Chemistry
 Pharmaceutical Intermediates
Pharmaceutical Intermediates
 Inhibitors/Agonists
Inhibitors/Agonists
 Material Science
Material Science







 For Research Only
For Research Only
 120K+ Compounds
120K+ Compounds
 Competitive Price
Competitive Price
 1-2 Day Shipping
1-2 Day Shipping