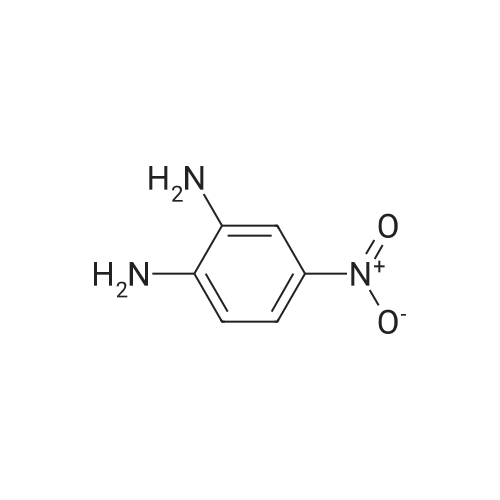
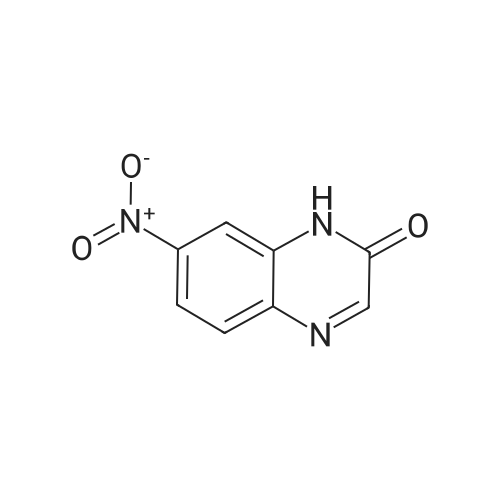
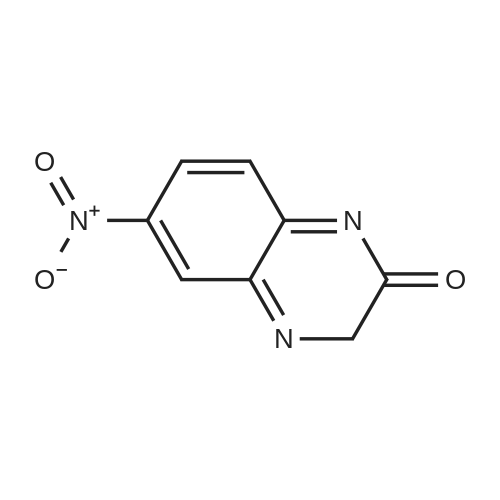
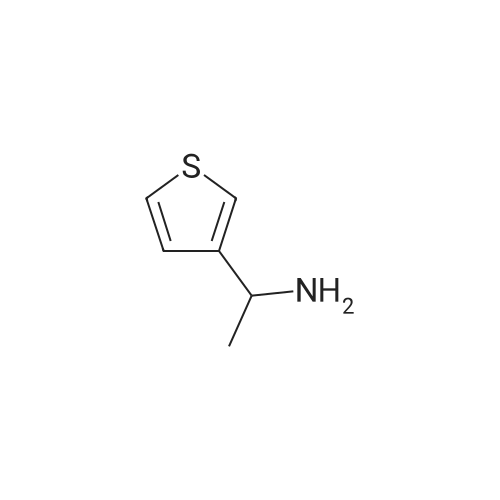
* All experimental methods are cited from the reference, please refer to the original source for details. We do not guarantee the accuracy of the content in the reference.
With sodium tetrahydroborate; In dimethoxyethane, 1,2; ethanol; at 50℃; for 7h;Heating / reflux;
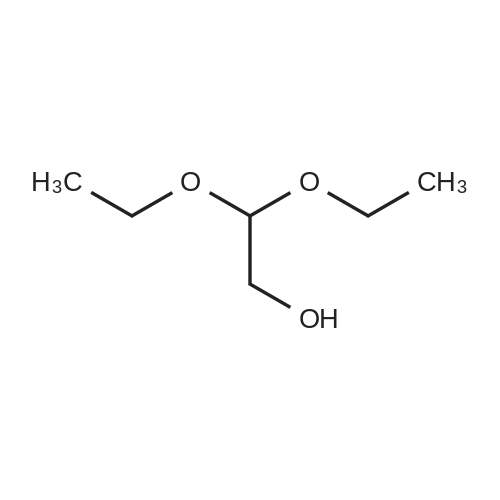
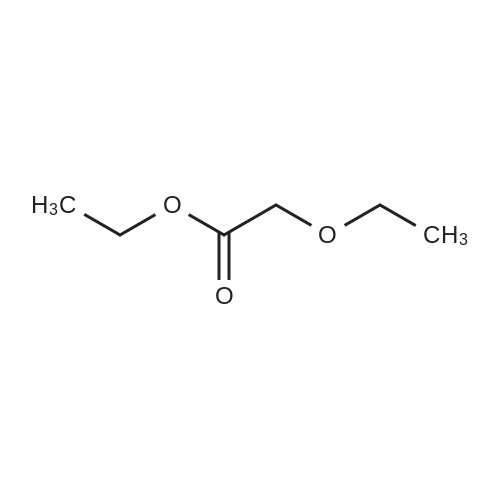
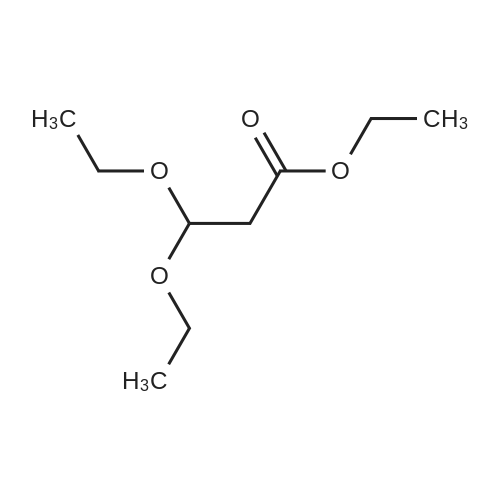
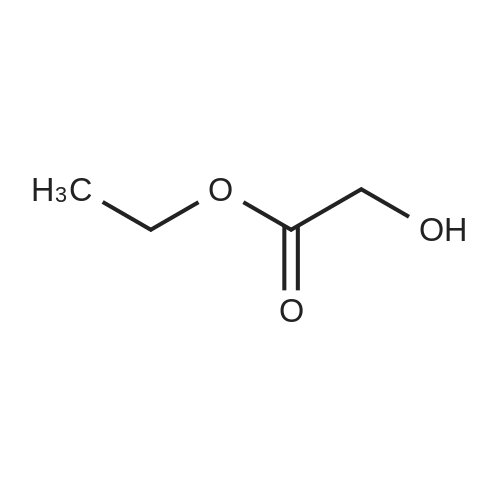
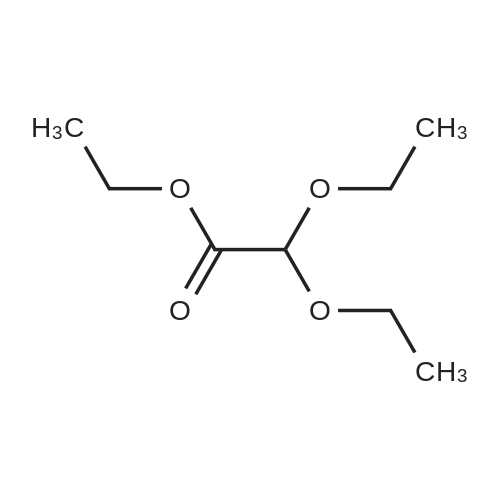
302,6 g (8 mol) of sodium borohydride was added to 2 1 of 1,2-dimethoxyethane (monoglyme) with stirring, after which 704,8 g (4 mol) ethyl diethoxyacetate dissolved in 4 1 of ethanol was added dropwise within 4 hours so that the temperature was kept below 50 °C. The mixture was then heated to reflux for 3 hours. Then 2 1 of ethanol was distilled off, and 4 1 of water was added dropwise while the remaining ethanol and then the 1,2-dimethoxyethane was removed by distillation. During the water addition an abundant precipitate was formed which dissolved towards the end of the addition. The mixture was cooled on an icebath and 600 g of potassium carbonate was dissolved therein while stirring. The mixture was extracted with 2 1 of diethyl ether and dried with MgSO4. The diethyl ether was evaporated off and the crude product was distilled in vacuo at 75-76 °C (15 mm Hg). Yield = 475.8 g = 88.7 percent [] Elemental analysis: CalculatedC 53.7percentH 10.5percentFoundC 53.11percentH 10.57percentIR: 3441 cm-1; 2976 cm-1; 2931 cm-1 ; 2883 cm-1; 1445 cm-1 1374 cm-1; 1345 cm-1; 1235 cm-1; 1134 cm-1; 1073 cm-1 (Between KBr plates)
(Z)-1-(3-chloro-4-fluorophenyl)-4,4-diethoxy-3-hydroxybut-2-en-1-one[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
35%
With lithium diisopropyl amide; In tetrahydrofuran; at -78 - 20℃; for 16h;
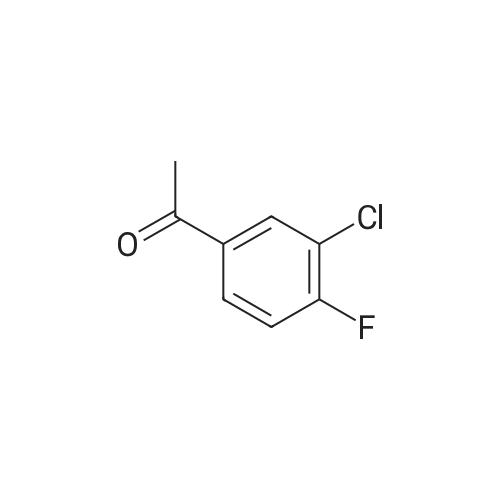
Intermediate A39A: (Z)-1-(3-Chloro-4-fluorophenyl)-4,4-diethoxy-3-hydroxybut-2-en-1-one To a -78 C. solution of <strong>[2923-66-2]1-(3-chloro-4-fluorophenyl)ethanone</strong> (16.25 g, 94 mmol) and ethyl 2,2-diethoxyacetate (20.73 mL, 113 mmol) in THF (392 mL) was added, dropwise, a solution of LDA (51.8 mL, 104 mmol, 2M in THF). The resultant reaction mixture was gradually allowed to reach room temperature and continued stirring for 16 h. The reaction was carefully quenched with water and diluted with EtOAc. The two layers were separated and the aq. layer was extracted with EtOAc (2*150 mL) The combined organic layer was washed with brine, dried over anhydrous MgSO4, filtered and the filtrate was concentrated under reduced pressure to provide a crude oil. It was purified by silica gel chromatography (1500 g Commodity column, eluting with a 10% EtOAc in hexanes). Fractions containing the product were combined and evaporated to afford Intermediate A39A (9.97 g, 35%) as a solid. MS(ES): m/z=257 [M-OEt]+.
With [methyl-3-(butyl-4-sulfonate) imidazolium]CuPW12O40; oxygen; at 159.84℃; under 6000.6 Torr; for 5h;
General procedure: In a typical process, 0.25 g lignin, 0.9 mmol POM-IL catalyst and 20 mL 100% ethanol werecharged into a 100 mL stainless autoclave (Andorra MED1220, Premex Co. Ltd.). After airpurging with pure oxygen five times and pressurizing to 0.8 MPa, the reactor was heated to thedesignated temperature and maintained for the desired time. Once the latter elapsed, the autoclavewas cooled rapidly to room temperature in an ice water bath. The reaction mixture was removedand the reactor was washed with anhydrous ethanol (3 5.0 mL). The IL catalyst was precipitatedat room temperature and used for the next run after drying (extra fresh catalyst was added tooffset transfer losses). The liquid mixture was then diluted by ethanol to 50 mL for qualitative andquantitative analysis, while dimethyl phthalate was used as the internal standard. When aqueoussolutions of ethanol were used, the spent mixture was rotary evaporated under reduced pressurefor solvent recovery. The concentrated liquor was esterified with 10 mL anhydrous ethanol at 373K for 2 h and then diluted to 50 mL with ethanol. Volatile products were qualitatively andquantitatively analyzed via gas chromatography-mass spectrometry (GC-MS) and gaschromatography-flame ionization detection (GC-FID). Residual lignin can be obtained throughsimple precipitation processes. Organosolv lignin was recovered as follows: 60 mL deionized water was added into 20 mL of the above reaction mixture causing precipitation. The mixture wasthen separated using centrifugation and was dried until a constant weight was obtained. For therecovery of dealkaline lignin the mixture obtained after reaction was acidified to pH=2 with 1.0mol L-1 HCl solution and the same procedure described for organosolv lignin was conducted.In the atmosphere investigation, a mixture of nitrogen and oxygen with various molar ratioswas used, while depolymerization of lignin was conducted at 433 K for 5.0 h in the single stageexperiments. For a typical two-stage process, the lignin was first depolymerized employing theaforementioned conditions. When the mixture was cooled to room temperature, an extra 0.8 MPanitrogen or oxygen was purged into the reactor and the reaction was heated to 433 K for 1.0 or 2.0h. The product separation and analysis procedure remained unchanged to that describedpreviously. In comparative and control experiments, a series of model compounds (monolignolsand potential intermediate products) were tested under the same procedures as that for lignin (i.e.,0.25 g model compound, 0.9 mmol POM-IL catalyst and 20 mL 100% ethanol solvent). Triplicateexperiments were conducted and the data shown in this study is the average.






 Chemistry
Chemistry
 Pharmaceutical Intermediates
Pharmaceutical Intermediates
 Inhibitors/Agonists
Inhibitors/Agonists
 Material Science
Material Science







 For Research Only
For Research Only
 120K+ Compounds
120K+ Compounds
 Competitive Price
Competitive Price
 1-2 Day Shipping
1-2 Day Shipping