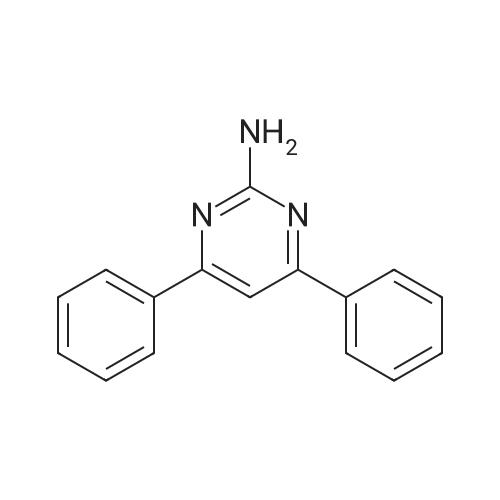
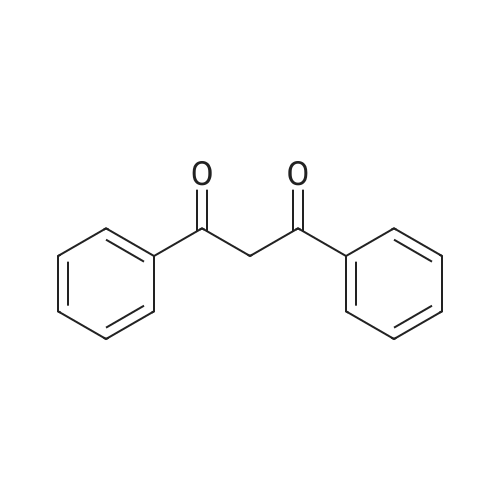
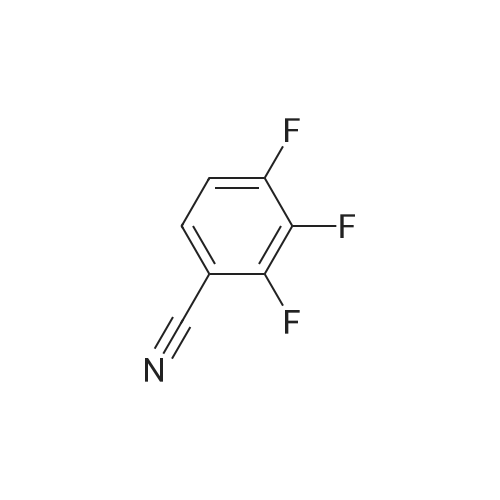
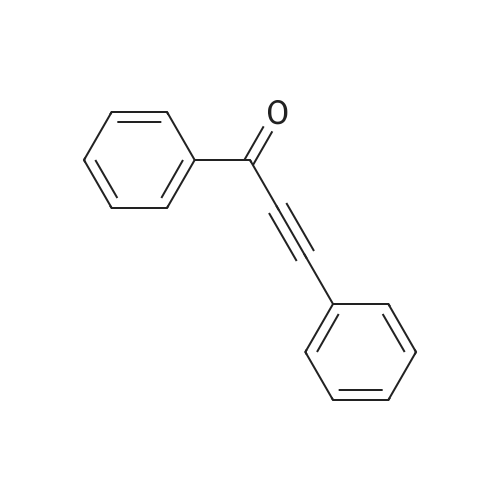
* All experimental methods are cited from the reference, please refer to the original source for details. We do not guarantee the accuracy of the content in the reference.
General procedure: To a suspension of guanidine carbonate (1.5-5 eq) in ethanol (2 mL/mmol) was added γ-aryl-β-ketoester (1.43-26.6 mmol), and the reaction mixture heated at 80 °C for 15-64 h. Followingreaction completion by TLC, the mixture was cooled to ambient temperature, filtered and thesolid triturated with water (5-20 mL) and acetone (5-20 mL) to give the title compound.
Reference:
[1] Tetrahedron, 2015, vol. 71, # 39, p. 7339 - 7343
[2] Journal of the American Chemical Society, 1998, vol. 120, # 27, p. 6761 - 6769
[3] Justus Liebigs Annalen der Chemie, 1891, vol. 262, p. 365
[4] Journal fuer Praktische Chemie (Leipzig), 1893, vol. <2> 47, p. 203
2
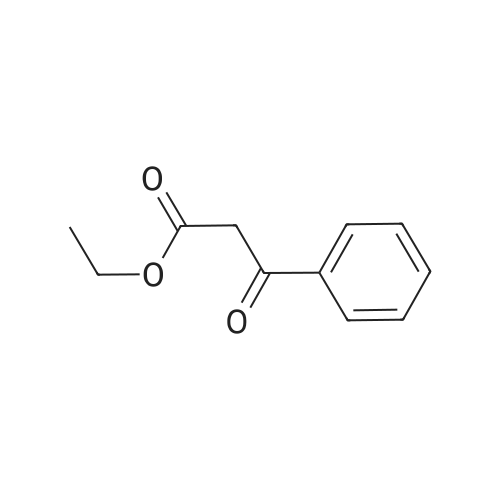
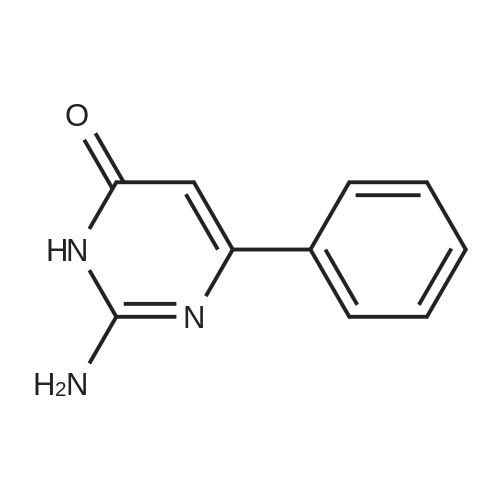
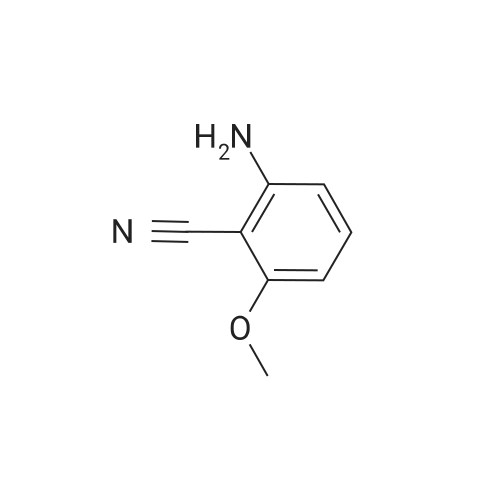
[ 593-85-1 ]
[ 118-92-3 ]
[ 20198-19-0 ]
Reference:
[1] Chemische Berichte, 1905, vol. 38, p. 1212[2] Chemische Berichte, 1910, vol. 43, p. 1021
3
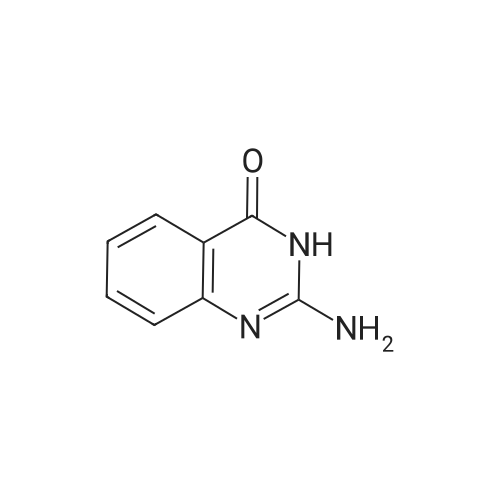
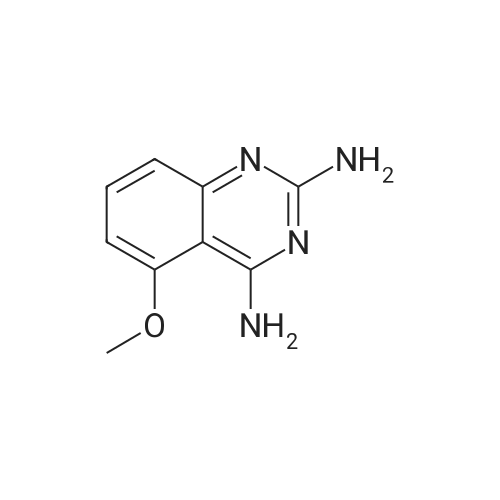
[ 1009734-34-2 ]
[ 593-85-1 ]
[ 1009734-33-1 ]
Reference:
[1] Journal of Medicinal Chemistry, 2008, vol. 51, # 3, p. 449 - 469
With sodium hydroxide; In butan-1-ol; at 120℃; for 2h;
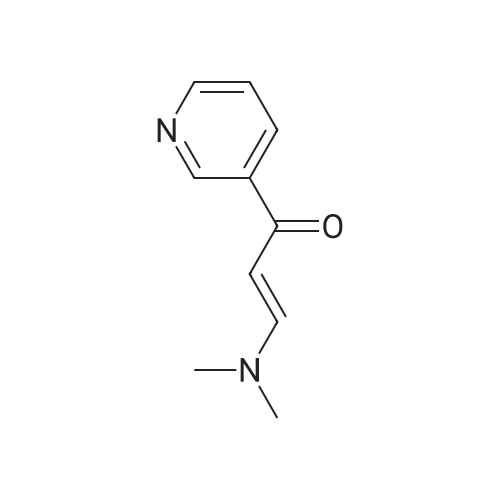
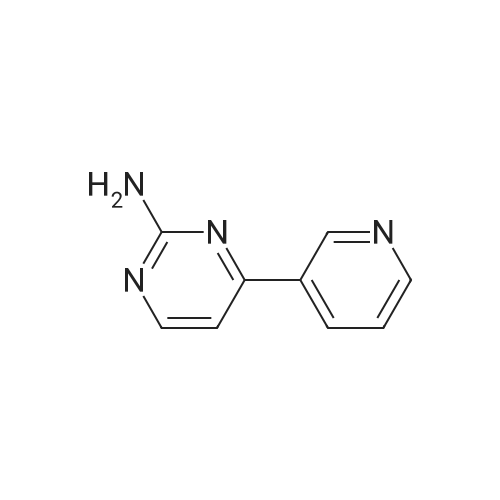
NaOH (0.78 g, 19.5 mmol) was added to a mixture of 3-(dimethylamino)-l-(pyridin-3- yl)prop-2-en-l-one (3.52 g, 20.0 mmol) and guanidine carbonate (1.80 g) in n-butanol (20 mL). The mixture was heated at 1200C for 2 h. After cooling, the precipitates formed was collected by filtration and dried under vacuum to afford the desired product (2.3g, 66percent). LCMS: (M+H)+= 173.2.
In iso-butanol; for 24h;Heating / reflux;
4-Pyridin-3 - yl-pyrimidin-2- ylamine (7)[0042] A mixture of 3-acetylpyridine (20.72 g, 171.0 mmol) and N, N-dimethylformamide dimethyl acetal (60 mL, 450 mmol) is heated at 110 0C. After reaction overnight, the reaction mixture is cooled to room temperature and concentrated. Ethyl ether (20 mL) and hexanes (60 <n="17"/>P A T E N TGNF Docket No.: P1286PC10mL) are added to the residue. The resulting solid is collected by filtration, washed with hexanes, and dried to afford 3-dimethylamino-l-pyridin-3-yl-propenone which is used for next reaction without further purification. A mixture of 3-dimethylamino-l-pyridin-3-yl-propenone (17.6 g, 100 mmol) and guanidine carbonate (10.8 g, 60.0 mmol) in 2-butanol (100 mL) is heated at reflux. After 24 hours, the reaction mixture is cooled to room temperature and concentrated. The residue is triturated in water (50 mL). The solid is collected by filtration, washed with water and dried to afford the desired product 4-pyridin-3-yl-pyrimidin-2-ylamine as a white solid. C29H31N7O Exact MS: 172.07. Found MS m/z 173.1 (M+l). 1H NMR (DMSO-J6): delta 9.23 (s, IH), 8.69 (m, IH), 8.37 (m, 2H), 7.54 (d, IH), 7.20 (d, IH), 6.80 (s, 2H).
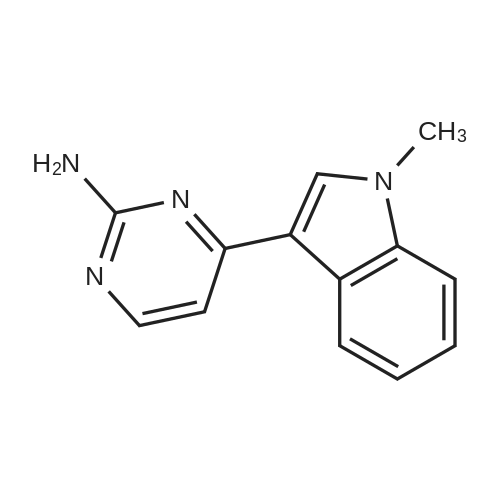
Preparation of I where R1 and R2 are Hydrogen, R3 is 1-methylindol-3-yl, and R4 and R5 are Hydrogen 3-Acetyl-1-methylindole (0.870 g) was dissolved in 3 ml of absolute ethanol. Tert-butoxybis(dimethylamino)methane (Bredereck's reagent) (0.960 g) in 3 ml of ethanol was added to this solution at reflux temperature. The solution was refluxed for 2 days and the solvent was removed at room temperature under vacuum. The residue was triturated with a 7:3 mixture of hexane/ethyl acetate to give a solid (0.094 g). The solid was mixed with guanidine carbonate (0.037 g) and the mixture was heated to 120C for 14 hours. The reaction mixture was dissolved in hot absolute ethyl alcohol, filtered, and recrystallized to give a white, crystalline solid of 2-amino-4-(1-methylindol-3-yl)-pyrimidine (0.039 g). Treatment of the crystalline solid with hydrochloric acid-ethyl alcohol and recrystallization of the salt from ethanol gave 2-amino-4-(1-methylindol-3-yl)-pyrimidine hydrochloride (0.0098 g), m.p. 274-276C.
With piperazine immobilized inside the mesochannels of magnetic MCM-41 as an organic base (a-Fe2O3-MCM-41-piperazine); In neat (no solvent); at 80℃; for 1h;Green chemistry;
General procedure: A mixture of benzaldehyde derivatives (1 mmol), acetophenone or 4-methylacetophenone(1 mmol), and benzamidine hydrochloride (1 mmol) or guanidiniumcarbonate (0.75 mmol) along with the catalyst (0.05 g) was thoroughly ground and then transferred into a reaction vessel where this mixture was stirred at 80 C for appropriate times. The reactions were monitored by thin-layer chromatography[TLC; ethyl acetate (EtOAc):petroleum ether, 1:4]. After completion of the reaction,the mixture was allowed to cool to room temperature, then chloroform (3 mL) wasadded and the mixture was stirred for an extra 30 min. After collecting the magneticcatalyst with an external magnet, the solvent was removed in vacuo and theprecipitates were recrystallized from ethanol to furnish the desired trisubstitutedpyrimidine derivatives.
benzyl 2-amino-4-hydroxy-5H,6H,7H-pyrrolo[3,4-d]pyrimidine-6-carboxylate[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
With triethylamine; In tert-butyl alcohol; at 80℃; for 24h;Inert atmosphere;
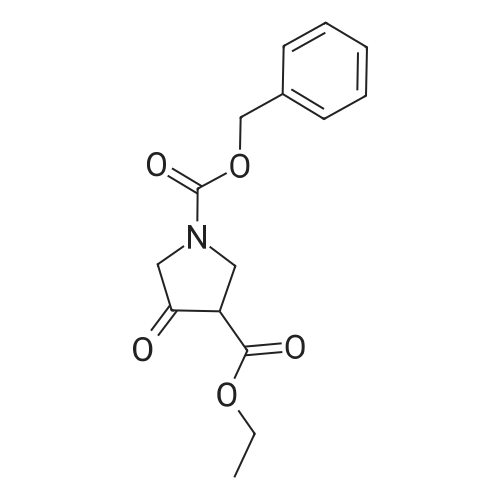
General procedure: Acetamidine hydrochloride (16.94 g, 179 mmol) was added to a solution of 1-benzyl 3- ethyl 4-oxopyrrolidine-l,3-dicarboxylate (CAS 51814-19-8) (29 g, 100 mmol) and triethylamine (25 mL, 179 mmol) in tert-BuOH (250 mL). The mixture was heated at 80 C for 24 hours under nitrogen. The mixture was concentrated in vacuo and partitioned between water and EtOAc. THF was added and the mixture filtered to give benzyl 4- hyckoxy-2-memyl-5H,6H,7H-pyrrolo[3,4-d]pyrimidine-6-carboxylate (11.18 g). The organic phase from the filtrate was passed through a pad of silica, eluting with EtOAc, and the resulting solution concentrated in vacuo. The residue was suspended in ether and THF (4:1, 200 mL) and filtered to afford the title compound. 1H NMR (400 MHz, DMSO-i): delta ppm 2.32 (s, 3H), 4.33 - 4.54 (m, 4H), 5.14 (s, 2H), 7.29 - 7.45 (m, 5H), 12.57 (br s, 1H). MS ES+ 286
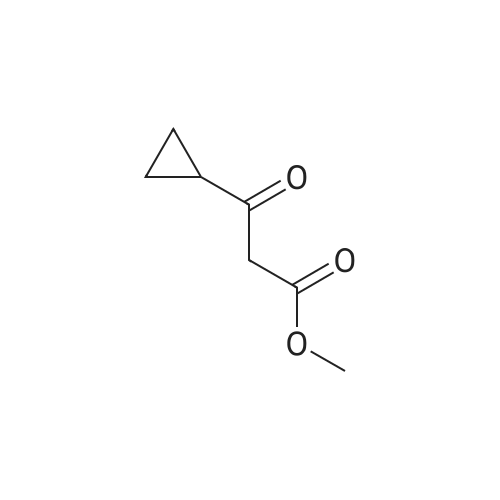
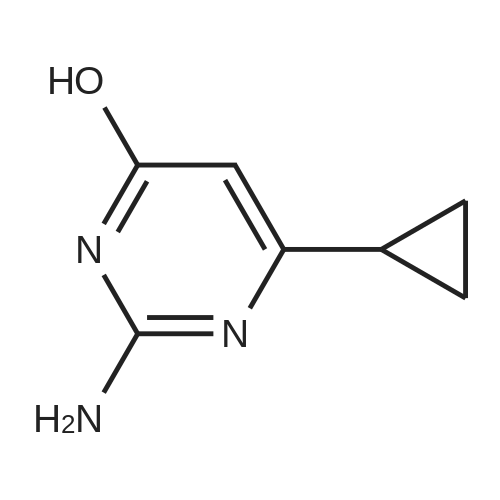
To a suspension of methyl 3 -cyclopropyl-3 -oxopropanoate (8.1 g, 57 mmol) and guanidine carbonate (5.65 g, 63 mmol) in ethanol (70 mL) at rt, was added NaOEt (7.75 g, 114 mmol) portion-wise over 20 min. After the addition, the resulting mixture was warmed to reflux and stirred overnight. After the reaction was complete as indicated by TLC analysis, the resulting suspension was concentrated under reduced pressure to remove most of the ethanol. The residue was diluted with a saturated NELCl solution (100 mL) and extracted with EA (3 x 200 mL). The combined organic layer was dried with Na?.S04 (50 g), filtered and concentrated to give 9 g of crude product. The crude product was slurried in a mixed solvent (Methanol/MTBE = 1/10, 33 mL) at rt overnight. After filtration and drying, 7.2 g of the title compound was obtained. LC-MS (Method A) (ESI+): m/z 152 (M+H)+; 1H-NMR (300 MHz, DMSO -d6) d 10.45 (s, 1H), 6.37 (s, 2H), 5.48 (s, 1H), 1.65 (m, 1H), 078-0.83 (m, 4H).






 Chemistry
Chemistry
 Pharmaceutical Intermediates
Pharmaceutical Intermediates
 Inhibitors/Agonists
Inhibitors/Agonists
 Material Science
Material Science







 For Research Only
For Research Only
 120K+ Compounds
120K+ Compounds
 Competitive Price
Competitive Price
 1-2 Day Shipping
1-2 Day Shipping