| 92% |
With sulfuric acid; at 60℃; for 6h;Inert atmosphere; Large scale; |
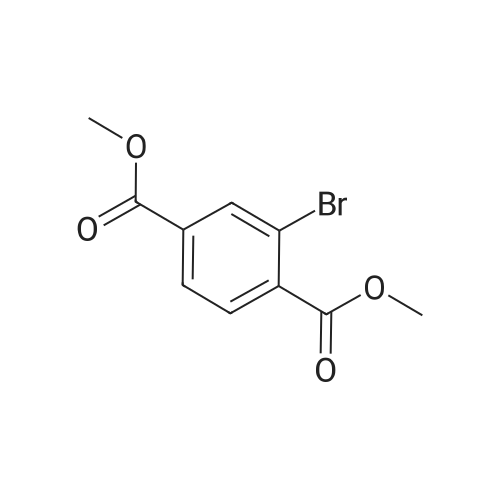
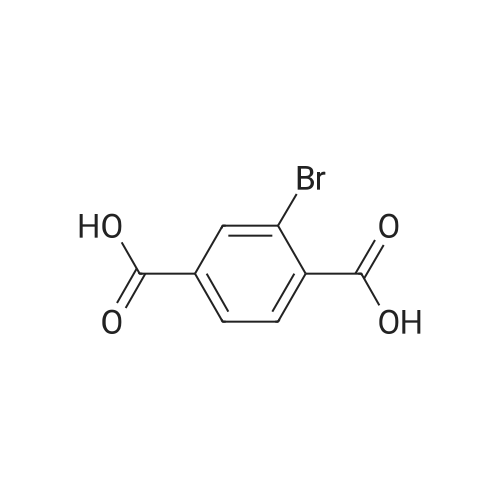
In nitrogen atmosphere, 2-bromoterephthalic acid (30.0 kg, 122.4mol) was suspened in methanol (95 kg), cooled to about 5 C, and 98 weight % sulfuric acid (33.0 kg) was added and the mixture was stirred at 60 C for 6 hours. TLC is confirmed for the end of reaction, the reaction mixture is then cooled to the room temperature, methyl tert-butyl ether (220.0 kg) was added and the organic layer was washed with water (180.0 kg), and NaHCO3 aqueous solution (8 weight %, 180.0 kg) and salt water (24 weight %, 180.0 kg) , dried using anhydrous magnesium sulfate (6.0 kg) , concentrated under reduced pressure to obtain the title compound as pale yellow crystals (30.40 kg, yield 92.0%). |
| 92.2% |
With sulfuric acid; In water; at 65℃; |
As shown in Scheme 1, MOF-ligand (see the Fig.S5 for the 1H and 13CNMR spectra of the MOF-ligand) and UiO-66-C≡CH could be synthesized by following a similar published procedure [28].Compound 1 to a solution of 2-bromoterephthalic acid (7.5 g,40 mmol) in 250 mL methanol, conc. H2SO4 (15 mL) was added slowly.Then being stirred at 65C overnight. After methanol was removed by evaporation, 300 mL ethyl acetate (EA) was added. The mixture was washed with K2CO3 solution (1 M) and dried with Na2SO4. Finally, the crude product 1 was purified by flask silica gel column chromatography(white solid 7.7 g, yield 92.2 %). 1H NMR (400 MHz, CDCl3) δ 8.30 (d, J=1.8 Hz, 1 H), 8.12 - 7.93 (m, 1 H), 7.81 (t, J =6.7 Hz, 1 H), 4.00 3.93(m, 6 H). 13C NMR (600 MHz, CDCl3) δ 167.96, 164.36, 137.47, 134.56,132.18, 129.76, 126.96, 122.04, 47.07. |
| 83% |
With thionyl chloride; at 0 - 20℃;Inert atmosphere; |
To a solution of 2-bromoterephthalic acid (2.0 g, 8.2 mmol) in MeOH (20 mL) at 0 00 underN2was added thionyl chloride (5.8 g, 49 mmol) dropwise. The resulting mixture was stirred atroom temperature overnight. The mixture was concentrated and the residue partitioned between DCM (20 mL) and saturated aqueous NaHCO3 (15 mL). The aqueous phase was extracted with DCM (3 x 10 mL). The combined organic fractions were washed with saturated aqueous NaHCO3 (3 x 10 mL), brine (3 x 10 mL), dried (Na2SO4), filtered and concentrated to give the title compound (1.85 g, 83%) as a white solid. 1H NMR (400 MHz,ODd3) 6 8.30 (d, J = 1.6 Hz, 1 H), 7.99 (dd, J = 8.0, 1.2 Hz, 1 H), 7.80 (d, J = 8.0 Hz, 1 H),3.95 (s, 3H), 3.94 (s, 3H); LCMS RT 2.63 mm; m/z 273,275 [M+H]+ |
| 44% |
With thionyl chloride; for 4h;Reflux; |
2-Bromoterephthalic acid (2.0 g, 8.2 mmol) was dissolved in 50.0 mL of methanol and 7.0 mL SOCl2. The mixture was heated to reflux for 4.0 h. After cooling to room temperature, MeOH was removed by evaporation and then 60 mL (20.0 mL * 3) of CH2Cl2 was added. The solution was washed water. Then the organic solution was dried with MgSO4. After solution was removed by evaporation, raw solid was obtained. Vacuum thermal sublimation and deposition was performed for a further purity (1.0 g, 0.44%). |
|
|
To a mixture of 2-bromoterephthalic acid (2.0 g, 8.2 mmol) and DMF (0.3 mL) in DCM (20 mL) was added oxalyl chloride (15.6 g, 123 mmol) and the mixture stirred at room (0570) temperature overnight. The reaction was then concentrated and the residue diluted with MeOH (50 mL) at 0 C and stirred for 10 min. The solvent was removed under reduced pressure to give the title product as a yellow oil that was used for the next step directly. LCMS: RT 2.61 min; m/z 273.0, 275.0 [M+H] +. |






 Chemistry
Chemistry
 Pharmaceutical Intermediates
Pharmaceutical Intermediates
 Inhibitors/Agonists
Inhibitors/Agonists
 Material Science
Material Science







 For Research Only
For Research Only
 120K+ Compounds
120K+ Compounds
 Competitive Price
Competitive Price
 1-2 Day Shipping
1-2 Day Shipping


UiO-66-Y hybrid nanomaterials for boosted photocatalytic hydrogen evolution.jpg)