|
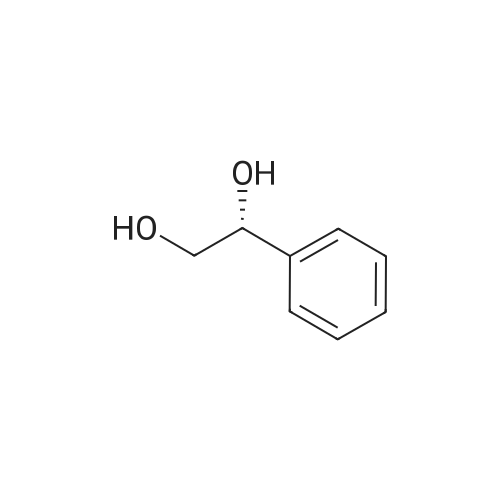
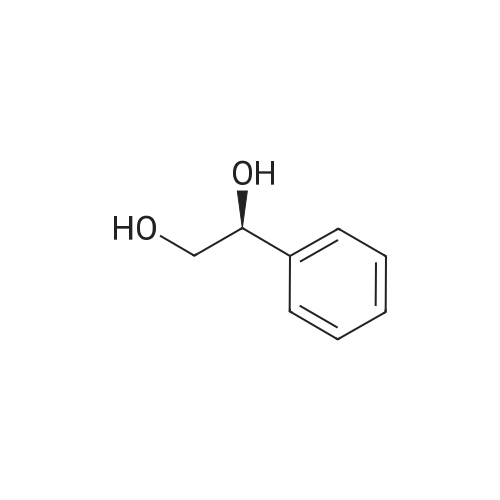
With potassium formate;(1,2,3,4,5-pentamethylcyclopentadienyl)IrCl[(S,S)-N-(p-toluenesulfonyl)-1,2-diphenylethylenediamine]; In water; toluene; at 50℃; for 24h;Product distribution / selectivity; |
The reaction was performed under the same conditions as those in Example B-1, except that 1.165 mg (1.6 μmol) of Cp*IrCl[(R,R)-TsDPEN] was used as the catalyst. HPLC analysis of the reactant confirmed that 1-phenyl-1,2-ethanediol with optical purity of 28% ee was produced in 30% yield. Comparison with Example B-1 demonstrated that it is superior to have a methyl group as the substituent on the sulfonyl group. |
|
With potassium formate;[(1,2,3,4,5-pentamethylcyclopentadienyl)IrCl((S,S)-N-(p-toluenesulfonyl)-1,2-cyclohexanediamine(1-))]; In water; toluene; at 50℃; for 24h;Product distribution / selectivity; |
The reaction was performed under the same conditions as those in Example B-1, except that 1.008 mg (1.6 μmol) of Cp*IrCl[(S,S)-TsCYDN] was used as the catalyst. HPLC analysis of the reactant confirmed that 1-phenyl-1,2-ethanediol with optical purity of 68% ee was produced in 10% yield. Comparison with Example B-1 demonstrated the superiority of MsDPEN as the dimaine ligand. |
|
With potassium formate;Cp*IrCl[(R,R)-(R)-CsDPEN]; In water; toluene; at 50℃; for 24h;Product distribution / selectivity; |
The reaction was performed under the same conditions as those in Example B-1, except that 1.467 mg (1.6 μmol) of Cp*IrCl[(R,R)-(R)-CsDPEN] was used as the catalyst. HPLC analysis of the reactant confirmed that 1-phenyl-1,2-ethanediol with optical purity of 87% ee was produced in 40% yield, showing that the catalytic efficiency of the iridium complex having camphorsulfonyl DPEN as the ligand is insufficient for the asymmetric reduction of ketones having a functional group. |
|
With hydrogen;Ru(trifluoromethanesulfonate)(N-(p-toluenesulfonyl)-1,2-diphenylethylenediamine)(η6-cymene); In methanol; at 50℃; under 76005.1 Torr; for 16h;Product distribution / selectivity; |
The hydrogenation reaction of α-hydroxyacetophenone was performed by the same method as in Example 30 except that the sulfonate catalyst was changed to Ru(OTf)[(S,S)-Tsdpen](p-cymene). As a result, 67% ee of optically active 1-phenyl-1,2-ethanediol was produced in a yield of only 3%. |
|
With potassium phosphate; Candida parapsilosis aldo-keto reductase CPAR5; NADPH; at 30℃; for 8h;pH 6.5;Enzymatic reaction; |
General procedure: Asymmetric reductions of various carbonyl compounds by the purified enzymes were carried out at 30C for 8h with mild shaking in a reaction mixture containing 0.1M potassium phosphate buffer (pH 6.5), 1gL-1 substrate, 10mM NADPH, and the purified enzyme of appropriate amount in a total volume of 2mL. In order to determine the absolute configuration of chiral alcohols, the reaction products were extracted with ethyl acetate or hexane and the organic layer was used for analysis. The optical purity of the reaction products were determined by chiral HPLC (HP 1100, Agilent, USA) equipped with Chiralcel OB-H column (4.6mm×250mm; Daicel Chemical Ind. Ltd., Japan) or chiral GC (7890A, Agilent, USA) equipped with FID detector and Chrompack Chirasil-Dex CB chiral capillary column (25m×0.25mm; Varian, USA) [21]. |
|
With silver tetrafluoroborate; diethoxymethylane; C26H29N3O2*Cl(1-)*Ir(1+)*C8H12; at 20℃; for 20h; |
General procedure: A flask was charged with azolium salt L12 (0.02 mmol, 9.1 mg),Ag2O (0.01 mmol, 2.4 mg) and CH2Cl2(1 mL). After stirring the resulting mixture at room temperature for 2 h in the dark, CH2Cl2 was removed in vacuo. Then, a THF (1 mL) solution of [IrCl(cod)]2(0.01 mmol, 6.9 mg) was added to the reaction vessel. The resulting mixture was stirred at room temperature for an additional 4 h in the dark, filtered through a membrane filter, and evaporated to dry-ness in vacuo. Subsequently, to the resulting flask containing yellow solid of the unpurified IrCl(cod)(NHC) complex, a solution of AgBF4(0.025 mmol, 4.9 mg) in CPME (2 mL) was added, and then stirred at room temperature for 1 h. Finally, propiophenone (0.5 mmol,66 mg) and (EtO)2MeSiH (2.25 mmol, 294 mg) were added to the resulting CPME solution (see Appendix A. Supplementary data fordetails). After stirring at room temperature for 20 h under open-air conditions, K2CO3(2 mg) and MeOH (2 mL) were added. Then, the resulting mixture was stirred at room temperature for 2 h. Afterevaporation of the solvents, the residue obtained was purified bycolumn chromatography on silica gel (Et2O/n-hexane = 3:7) to give(S)-1-phenyl-1-propanol (61 mg, 91% isolated yield). The ee was measured by chiral GLC. |
|
With C. parapsilosis strains containing exogenous gene scrII; In aq. phosphate buffer; at 35℃; for 45h;pH 6.0;Microbiological reaction; |
methodsThe asymmetric reduction of 2-HAP to (S)-PED by C. parapsilosiswas conducted as described by Nie et al. [26] with some mod-ifications. After 36 h incubation, cells were collected at 6000 × gcentrifugation and washed twice with saline. For asymmetric reac-tion, 1 mL of the reaction mixture contained 0.2 M acetate (pH4.0-6.0) or 0.2 M phosphate buffer (pH 6.0-7.0), 2-HAP (1 g/L to30 g/L) and 10% (W/V) wet-cells of C. parapsilosis. The productswere extracted with ethyl acetate, and the optical purity andyield were determined by HPLC on a Chiralcel OB-H column (Dai-cel Chemical Ind, Ltd., Japan) as described previously [26]. Theoptical purity and yield were calculated basing on the followingequations |
|
|
The N. corallina cells were incubated in 50 mL phosphate buffer (0.1 mol L-1, pH 7.00),30 min, at 28-30 C in an orbital shaker (150 rpm), and 2a or 2b was added to the whole cellsat a substrate:cells mass ratio of 1:100 for 2a and 1:400 for 2b, using 0.6 vol. % of N,N--dimethylformamide and shaken under the same conditions. The experiments were performedin triplicate at different final pH values of the culture media and times. The sample was centrifugedat 4500 rpm for 15 min and then extracted with ethyl acetate (4×15 mL), and the organiclayer was concentrated to dryness. The product was dissolved in 0.5 mL of HPLC grade2-propanol. The GC analysis was performed with a HP-5 column (30 m×0.33 mm, 0.25 μm)(Hewlett-Packard, Germany) at 80-200 C, with N2 as the carrier gas at 1.0 mL min-1. Theoven temperature was ramped from 80-200 C at 10 C min-1, held for 3 min, decreased to 80C at 25 C min-1 and held for 2 min. The retention times were tR(2a) = 4.90 min and tR(2b) == 5.79 min. Then, the analyse was realized by HPLC using a chiracel OB-H (25.0 cm×0.46cm, 0.5 μm) column (Daicel Chemical Industries, Tokyo, Japan). The mobile phase washexane:2-propanol (90:10), 0.5 mL min-1, λ = 220 nm, 24 C. For the reduction of 2a, theretention times were tR(R)-2b = 15.10 min, tR(S)-2b= 18.90 min and tR(2a) = 28.90 min. Forthe oxidation of 2b, the mobile phase was hexane:2-propanol (90:10), 0.8 mL min-1, λ = 260nm, 24 C. The retention times were tR(R)-2b = 9.04 min, tR(S)-2b = 11.38 min and tR(2a) == 17.12 min, The absolute configuration of 2b was assigned according to the literature.23 |






 Chemistry
Chemistry
 Pharmaceutical Intermediates
Pharmaceutical Intermediates
 Inhibitors/Agonists
Inhibitors/Agonists
 Material Science
Material Science







 For Research Only
For Research Only
 120K+ Compounds
120K+ Compounds
 Competitive Price
Competitive Price
 1-2 Day Shipping
1-2 Day Shipping