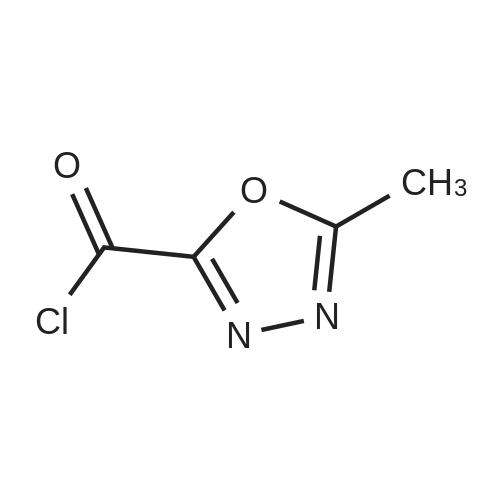
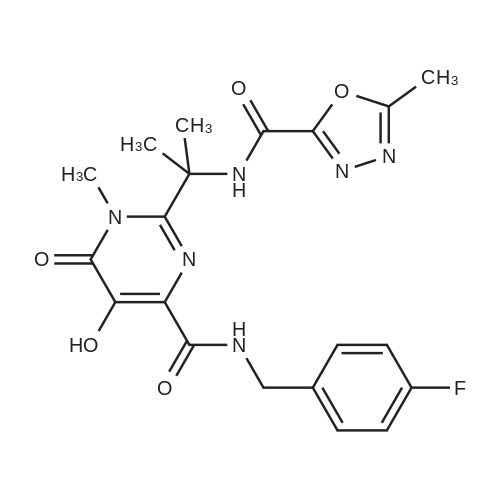
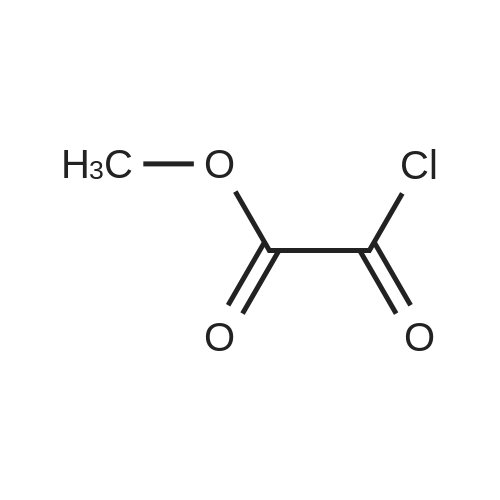
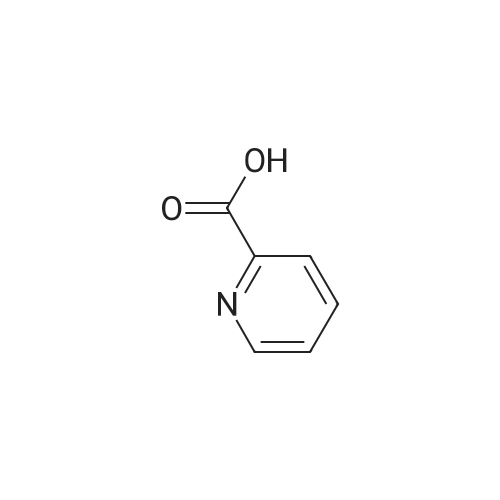
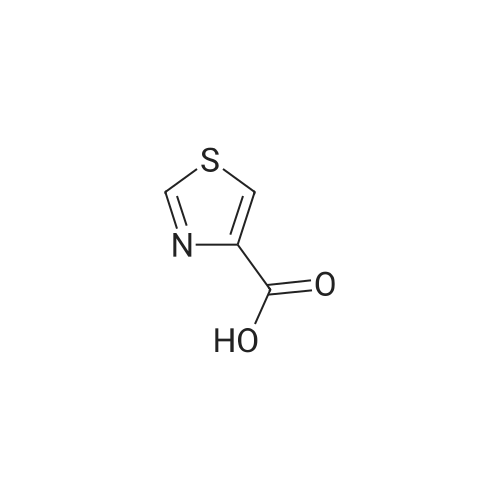
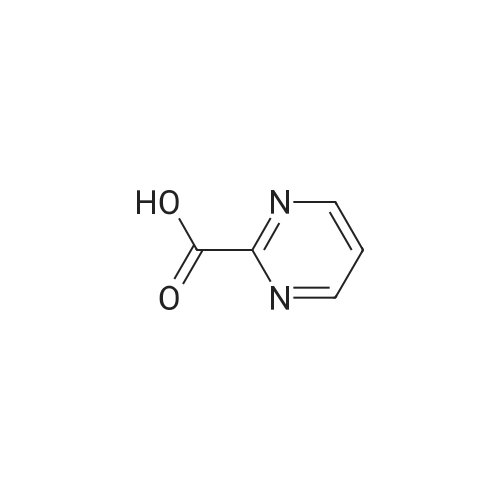
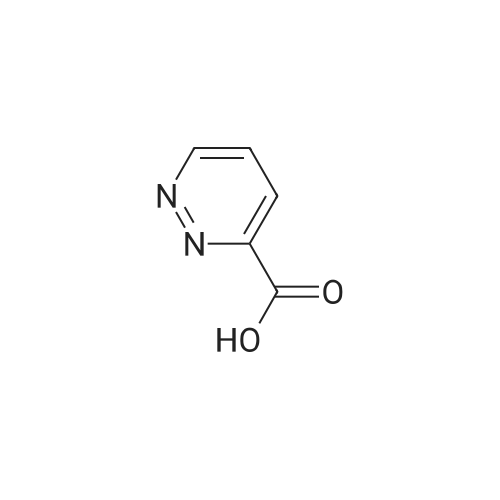
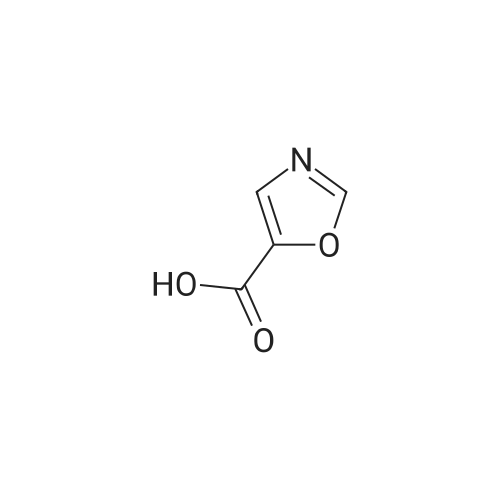
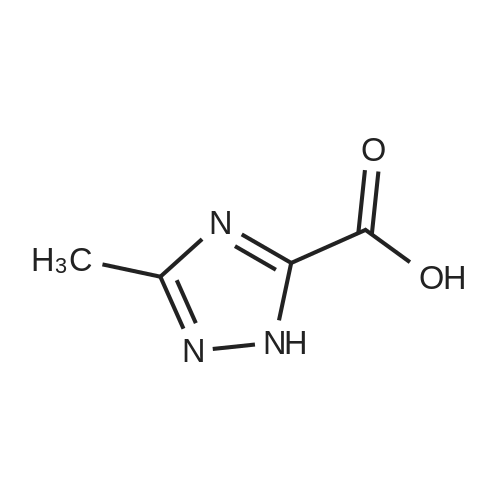
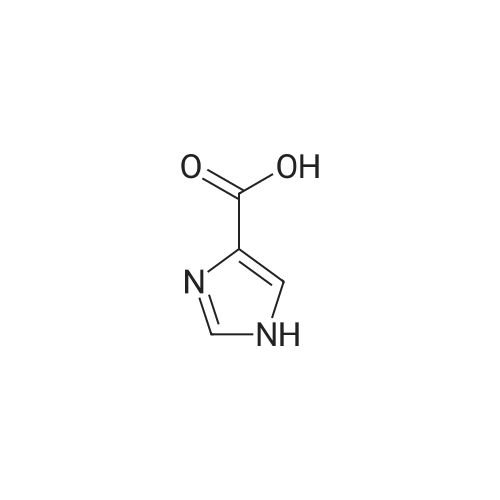
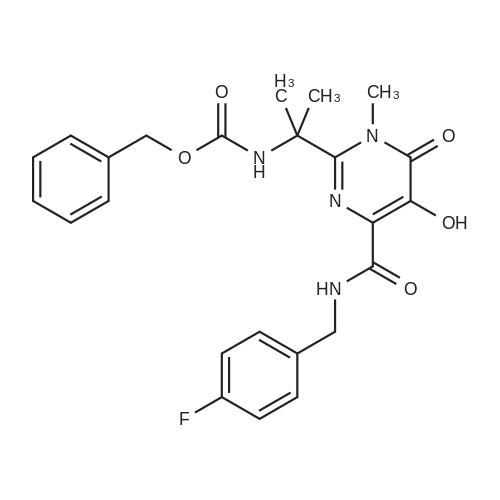
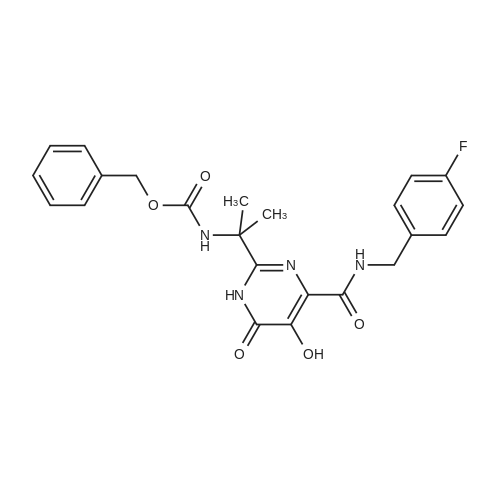
| 96% |
With methanesulfonic acid; hydrogen;5% palladium over charcoal; In methanol; at 50℃; under 2068.65 Torr; for 3 - 4h; |
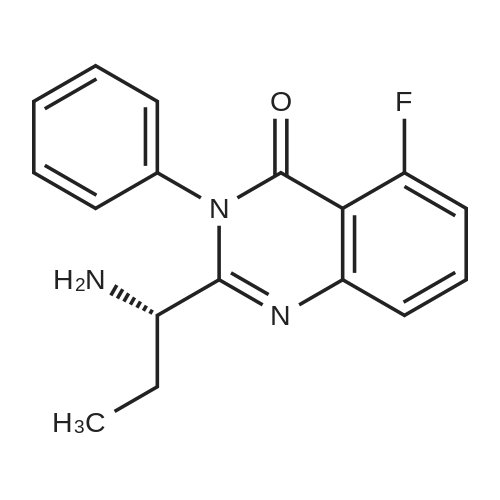
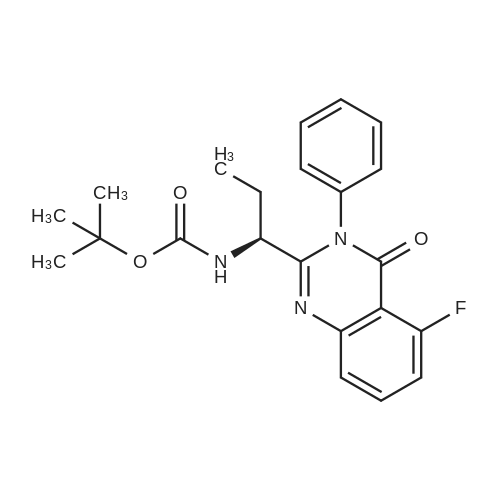
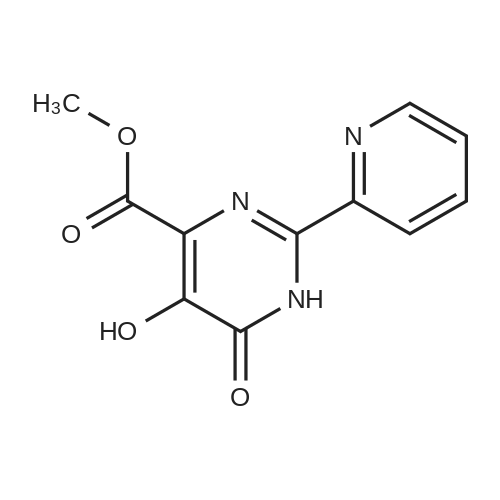
Hvdrogenation of Cbz-amide[MW = 468.48] [MW = 334.35]Material MW mmoles Mass VolumeCBz amide (g) 468.48 21.33 1O gMeOH 8O mL5% Pd/C (50% wet) 0.15 gMSA 96.1 22.4 1.45 mL water 8 mL cake wash (4: 1 MeOH:H2theta 2O mL1 N NaOH 22.4 22.4 mL final cake wash (water) 3O mLA stainless steel hydrogenation vessel was preconditioned with MeOH, Pd/C catalyst and MSA under the reaction conditions described below. Cbz-amide g (IOg) was then slurried in MeOH (80 mL) in the preconditioned vessel. MSA (1.45 mL) was added to the slurry in one portion at room temperature. 5% Pd/C (0.15g, 50% wet) was also added to the hydrogenation vessel. Hydrogen was charged to the vessel in three successive vacuum/hydrogen purge cycles, after which the mixture was hydrogenated at 40 psig for 3-4 hour at 500C. Following hydrogenation, water (8 mL) was added to the reaction mixture, the mixture was stirred, and the catalyst was filtered and washed with 4:1 MeOH:water (20 mL). The pH of combined filtrates was adjusted to pH 7 to 8.0 by slow addition of 1 N NaOH (22.4 mL), which precipitated a solid. The slurry was stirred at 0-5 0C for 4 hours and the solid filtered, washed with water (30 mL), collected and dried in vacuo at 500C. The product amine (as hydrate) was obtained as a white crystalline solid (7.7g) in 96% yield (corrected for KF), 89%LCWP, 99.8% LCAP, JCF=I l wt.%HPLC Method A (product assay): column: 25 cm x 4.6 mm Zorbax RX-C8; mobile phase: A =0.1% H3PO4, B = CH3CN, 0 minutes (80% A/ 20% B), 20 minutes (20% A/ 80% B), 25 minutes (20% AJ EPO <DP n="35"/>80% B); flow: 1.0 mL/minute; wavelength: 210 nm; column temperature: 40 0C; retention times: des- fluoroamine byproduct - 5.5 min, amine product - 5.85 minutes, toluene - 16.5 minutes, Cbz-amide - 16.82 minutes.HPLC Method B (product purity): column: 25 cm x 4.6 mm YMC-basic; mobile phase: A =25 mmol KH2PO4 adjusted to pH=6.1, B = CH3CN, 0 minutes (90% A/ 10% B), 30 minutes (30% A/ 70% B), 35 minutes (30% A/ 70% B); flow: 1 mL/minute; wavelength: 210nm; column temperature: 30 0C; retention times: des-fluoroamine - 9.1 minutes, amine - 10.1 minutes, toluene - 24.2 minutes, Cbz amide - 25.7 minutes |
| 96% - 99.8%Chromat. |
With methanesulfonic acid; hydrogen;5% palladium over charcoal; In methanol; at 20 - 50℃; under 2828.7 Torr; for 3 - 4h; |
A stainless steel hydrogenation vessel was preconditioned with MeOH, Pd/C catalyst and MSA under the reaction conditions described below. Cbz-amide g (1Og) was then slurried in MeOH (80 mL) in the preconditioned vessel. MSA (1.45 mL) was added to the slurry in one portion at room temperature. 5% Pd/C (0.15g, 50% wet) was also added to the hydrogenation vessel. Hydrogen was charged to the vessel in three successive vacuum/hydrogen purge cycles, after which the mixture was hydrogenated at 40 psig for 3-4 hour at 50C. Following hydrogenation, water (8 mL) was added to the reaction mixture, the mixture was stirred, and the catalyst was filtered and washed with 4: 1 MeOHrwater (20 mL). The pH of combined filtrates was adjusted to pH 7 to 8.0 by slow addition of 1 N NaOH (22.4 mL), which precipitated a solid. The slurry was stirred at 0-5 0C for 4 hours and the solid filtered, washed with water (30 mL), collected and dried in vacuo at 500C. The product amine (as hydrate) was obtained as a white crystalline solid (J. Ig) in 96% yield (corrected for KF), 89%LCWP, 99.8% LCAP, KF=Il wt.%. |
| 96% |
|
A stainless steel hydrogenation vessel was preconditioned with MeOH, Pd/C catalyst and MSA under the reaction conditions described below. Cbz-amide g (1Og) was then slurried in MeOH (80 mL) in the preconditioned vessel. MSA (1.45 mL) was added to the slurry in one portion at room temperature. 5% Pd/C (0.15g, 50% wet) was also added to the hydrogenation vessel. Hydrogen was charged to the vessel in three successive vacuum/hydrogen purge cycles, after which the mixture was hydrogenated at 40 psig for 3-4 hour at 500C. Following hydrogenation, water (8 mL) was added to the reaction mixture, the mixture was stirred, and the catalyst was filtered and washed with 4:1 MeOH:water (20 mL). The pH of combined filtrates was adjusted to pH 7 to 8.0 by slow addition of 1 N NaOH (22.4 mL), which precipitated a solid. The slurry was stirred at 0-5 0C for 4 hours and the solid filtered, washed with water (30 mL), collected and dried in vacuo at 500C. The product amine (as hydrate) was obtained as a white crystalline solid (7.7g) in 96% yield (corrected for KJ), 89%LCWP, 99.8% LCAP, KF=I 1 wt.%HPLC Method A (product assay): column: 25 cm x 4.6 mm Zorbax RX-C8; mobile phase: A =0.1% H3PO4, B = CH3CN, 0 minutes (80% A/ 20% B), 20 minutes (20% A/ 80% B)5 25 minutes (20% A/80% B); flow: 1.0 mL/minute; wavelength: 210 nm; column temperature: 40 0C; retention times: des- fluoroamine byproduct - 5.5 min, amine product - 5.85 minutes, toluene - 16.5 minutes, Cbz-amide - 16.82 minutes.HPLC Method B (product purity): column: 25 cm x 4.6 mm YMC-basic; mobile phase: A =25 mmol KH2PO4 adjusted to pH=6.1, B = CH3CN, 0 minutes (90% AJ 10% B), 30 minutes (30% A/ 70% B), 35 EPO <DP n="36"/>minutes (30% AJ 70% B); flow: 1 mL/minute; wavelength: 210nm; column temperature: 30 0C; retention times: des-fluoroamine - 9.1 minutes, amine - 10.1 minutes, toluene - 24.2 minutes, Cbz amide - 25.7 minutes. |
|
With hydrogen bromide; In water; at 28 - 62℃; for 2.5h; |
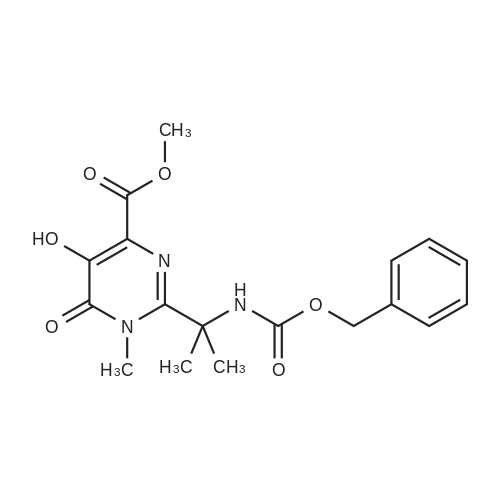
A mass of 730 g of aqueous hydrobromic acid was added to 100 g of benzyl (2- { 4-ft4-fluorobenzyl) carbamoyl]-5 -hydroxy- 1 -methyl-6-oxo- 1 ,6-dihydropyrimidin-2-yl}propan-2-yl)-carbamate at 28 + 3 C and stirred for 30 minutes. The reaction mixture temperature was raised to 62 + 3 C and stirring continued for 2 hours. After completion of the reaction, the reaction mixture was cooled to 28 ± 3 C and 300 mL of purified water was added. The reaction mixture was stirred and cooled to 12 + 3C. The pH of the reaction mixture was adjusted to 7.0 - 8.0 with a sodium hydroxide solution and further cooled to 3 ± 2 C. The reaction mixture was stirred for about 3- 4 hrs. The product was filtered then washed with water. Methanol (500 mL) was added to the wet material at 28 ±3 C and stirred for 1 hour to obtain a solid. The solid was filtered then washed with methanol. The product was dried under vacuumat 55 ± 3 C to get 2-(2-amino propan-2-yl)-N-(4-fluorobenzyl)-5-hydroxy-1-methyl-6-oxo- 1, 6-dihydro pyrimidine-4-carboxamide (Formula III). |
| 65 g |
With glycolic Acid; 5%-palladium/activated carbon; hydrogen; In methanol; under 6723.1 Torr; |
A slurry suspension of 100 g (0.213 mol) Benzyl (2-(4-((4-fluorobenzyl)carbamoyl)- 5-hydroxy-l-methyl-6-oxo-l,6-dihydropyrimidin-2-yl)propan-2yl)carbamate IVa, 38 ml (0.426 mol) glycolic acid (67% w/w) and 5g 5% Pd/C (50% w/w) in 3L methanol was hydrogenated at 130 psi for 3-4 h (>99.6% purity). The reaction mixture was filtered through celite bed and washed with 100 ml methanol. The combined filtrate was concentrated to a total volume of 1.1L at 25-28 C and neutralized by 65 ml triethylamine to pH= 9-10 at 25-28 C. The formed crystalline solid is collected by filtration, rinsed with 100ml methanol and dried under vacuum to afford 65 g (0.194 mol) of free amine (lib). (0147) 1H-NMR (500 MHz, DMSO-d6) delta 10.16 (br, 1 H), 7.33-7.30 (m, 2H), 7.14-7.10 (m, 2 H), 4.45 (d, J = 6.0 Hz, 2 H), 3.68 (s, 3 H), 1.56 (s, 6 H). (0148) 13C-NMR (125 MHz, DMSO-d6) delta 168.1, 162.1, 160.2, 135.6, 129.3, 123.2, 115.0, 55.9, 48.6, 41.3, 33.1, 28.2. |
| 3 g |
With sodium hydroxide; In ethanol; butan-1-ol;Reflux; |
In a 250 mL four necked round bottom flask added 5 g of compound 6 portion-wise to a alcoholic solution of sodium hydroxide (2 g) in n-butanol (20 mL) at room temperature under vigorous stirring. The slurry solution was slowly heated to reflux and maintain for 3-4 h until it TLC complies. The resulting brown colored precipitated solution was cooled to 20-25 C diluted with 50 mL of water to get clear solution. The obtained off-white coloured solid was filtered and dried after reaction mass pH was adjusted to 7.5 with conc. HCl (5 mL) to afford 3 g of compound 5 with HPLC purity 99.4 %. 1H NMR (DMSO-d6): delta 10.55 (s, 1H), 7.27-7.30 (m, 2H), 7.10(t, 2H), 4.43 (d, 2H), 3.60 (s, 3H), 1.58 (s, 6H); 13C NMR(DMSO-d6):167.87, 162.50, 159.95, 152.15, 145.17, 135.77,129.23, 122.92, 115.10, 56.02, 41.33, 33.01, 27.24. Mass: M+:334.35 , M+H: 435.1 (+ve Scan), M-H: 333.1 (-ve scan). |






 Chemistry
Chemistry
 Pharmaceutical Intermediates
Pharmaceutical Intermediates
 Inhibitors/Agonists
Inhibitors/Agonists
 Material Science
Material Science







 For Research Only
For Research Only
 120K+ Compounds
120K+ Compounds
 Competitive Price
Competitive Price
 1-2 Day Shipping
1-2 Day Shipping