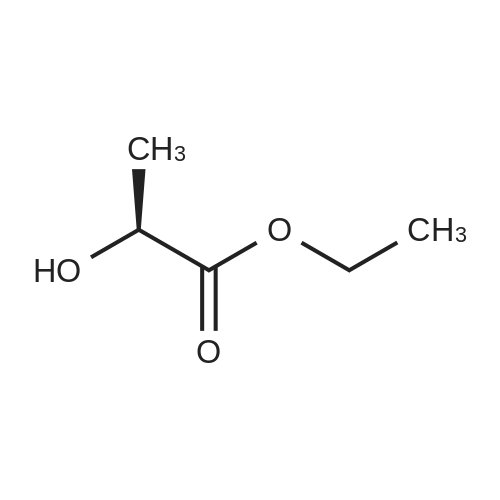
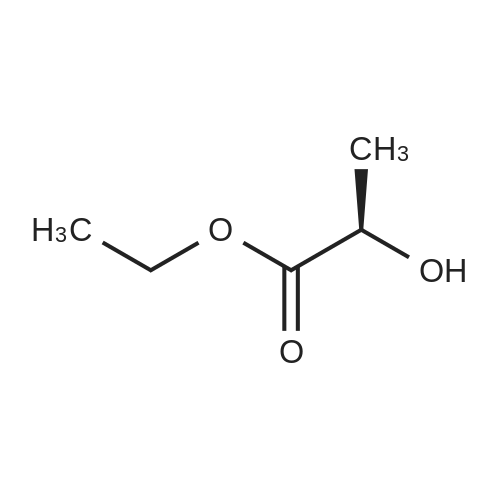
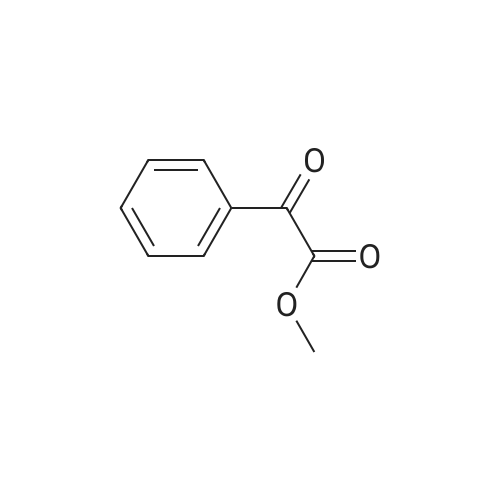
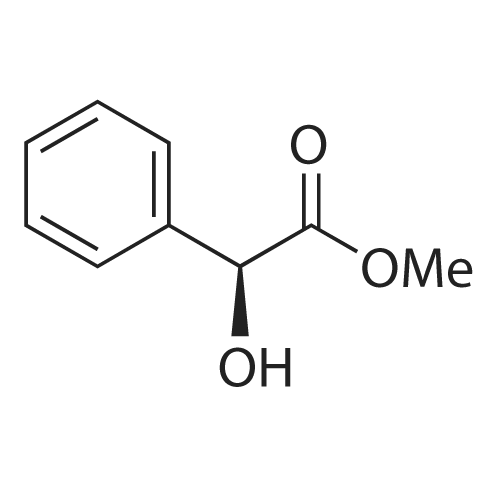
|
|
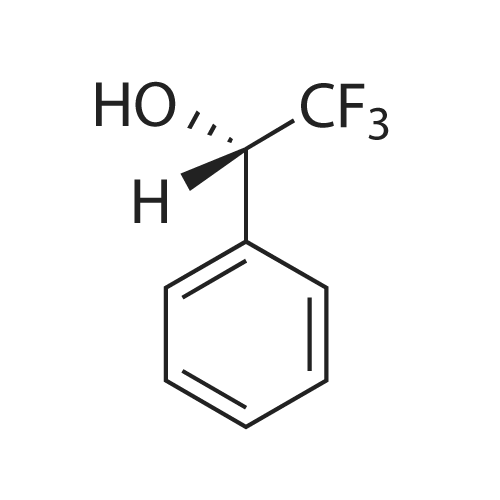
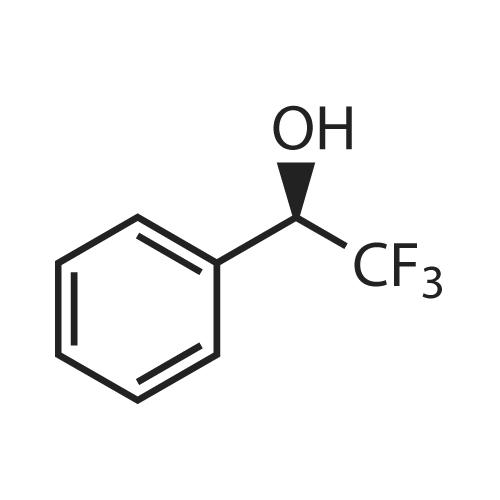
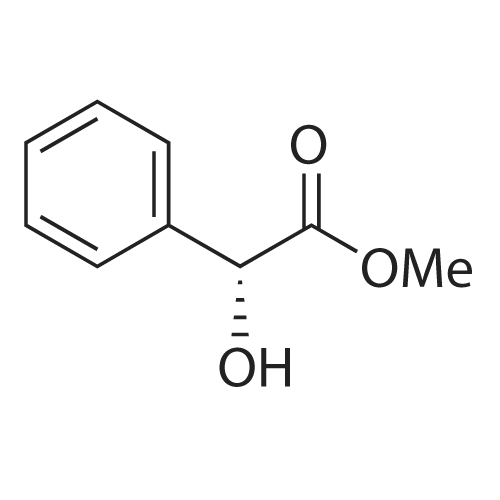
General procedure: To a solution of chiral lactam alcohol 2 (10.7 mg, 0.04 mmol, 10 mol %) in CHCl3 (0.8 ml) was added a 1 M BH3-THF solution (0.48 ml, 0.48 mmol). The mixture was stirred under Ar at room temperature for 10 min. A solution of trifluoroacetophenone (56 μl, 0.4 mmol) in CHCl3 (0.6 ml) was added dropwise over 1 h using a syringe pump. The reaction mixture was stirred until the ketone disappeared based on TLC (1 h). The reaction was quenched with MeOH (200 μl), extracted with ether and dried over MgSO4. Flash-chromatography of the crude mixture (hexane/AcOEt, 5:1) gave (S)-2,2,2-trifluoro-1-phenylethanol (63.4 mg, 90%). The ee was determined to be 83% by HPLC analysis using a Chiralcel OD column, hexane/i-PrOH = 97:3, 1.0 ml/min, and the absolute configuration was established by comparing the retention time of the commercially available (S)-2,2,2-trifluoro-1-phenylethanol [retention times = 22 and 27 min for (S) and (R), respectively]. |
|
|
General procedure: To a solution of chiral lactam alcohol 2 (10.7 mg, 0.04 mmol, 10 mol %) in CHCl3 (0.8 ml) was added a 1 M BH3-THF solution (0.48 ml, 0.48 mmol). The mixture was stirred under Ar at room temperature for 10 min. A solution of trifluoroacetophenone (56 μl, 0.4 mmol) in CHCl3 (0.6 ml) was added dropwise over 1 h using a syringe pump. The reaction mixture was stirred until the ketone disappeared based on TLC (1 h). The reaction was quenched with MeOH (200 μl), extracted with ether and dried over MgSO4. Flash-chromatography of the crude mixture (hexane/AcOEt, 5:1) gave (S)-2,2,2-trifluoro-1-phenylethanol (63.4 mg, 90%). The ee was determined to be 83% by HPLC analysis using a Chiralcel OD column, hexane/i-PrOH = 97:3, 1.0 ml/min, and the absolute configuration was established by comparing the retention time of the commercially available (S)-2,2,2-trifluoro-1-phenylethanol [retention times = 22 and 27 min for (S) and (R), respectively]. |
|
With 5 wt% platinum/alumina; hydrogen; trifluoroacetic acid; (8R,9S)-cinchonine; In toluene; at 0℃; under 7500.75 Torr; |
3.5 mmol 4, 48 mg 5 wt.% Pt/Al2O3, 6.8 μmol modifier in 10 mL toluene to which 3 μL trifluoroacetic acid was added, at 0 C and 10 bar. |
|
With aluminum oxide; hydrogen; 8R,9S-cinchoine; platinum; In N,N-dimethyl-formamide; at 20℃; under 750.075 Torr; for 1h; |
General procedure: enantioselective hydrogenation of 1 was carried out at room temperature and under atmospheric pressure, using a magnetically stirred (750 rpm) 25 ml glass reactor equipped with a rubber septum for sampling, vertical condenser and an entrance for the gas flow. Under standard conditions 3.7 mmol of 1 was dissolved in 5 ml of solvent together with 3.4 mol of CN. Finally, 21±1 mg of the activated catalyst was added to the reaction mixture and the system was flushed with nitrogen at room temperature |
|
With aluminum oxide; hydrogen; 8R,9S-cinchoine; platinum; In toluene; at 20℃; under 750.075 Torr; for 1h; |
General procedure: enantioselective hydrogenation of 1 was carried out at room temperature and under atmospheric pressure, using a magnetically stirred (750 rpm) 25 ml glass reactor equipped with a rubber septum for sampling, vertical condenser and an entrance for the gas flow. Under standard conditions 3.7 mmol of 1 was dissolved in 5 ml of solvent together with 3.4 mol of CN. Finally, 21±1 mg of the activated catalyst was added to the reaction mixture and the system was flushed with nitrogen at room temperature |
|
With dimethylsulfide borane complex; 2-hydroxy-N-[(2S)-1-hydroxy-3-phenylpropan-2-yl]benzamide; In tetrahydrofuran; at 65℃;Inert atmosphere; |
General procedure: A 25mL two-necked flask was charged with β-hydroxyamide 1-7 (0.05mmol, 10%) in dry and fresh THF (3mL), equipped with a magnetic stirrer and a connection to the combined nitrogen/vacuum line and closed with a septum. The air in the flask was replaced by nitrogen. β-hydroxyamides 1-7 were dissolved in THF (3mL) with stirring and a solution of BH3·SMe2 (0.5mmol, 10M) complex was added at 0C by a syringe. After the mixture was stirred for 1h at 65C, the freshly distilled ketone (0.5mmol) in freshly dried THF (2mL) was added over a period of 1.5h by a syringe at the same temperature. The reaction mixture was kept stirring at the 65C until the ketone was completely consumed. After stirring for a further 30min at rt, the reaction mixture was quenched by the addition of MeOH (2mL) and extracted with CH2Cl2 three times. The combined organic extracts were washed with brine and dried over MgSO4. After evaporating the solvent under reduced pressure, the product was purified by column chromatography on silica gel using petroleum ether/EtOAc (5:1; for 4′-nitroacetophenone: 5/3) as the eluent. The ee value was determined by HPLC with Chiralcel AS-3 or Chiralcel OD-H columns. |
|
With dimethylsulfide borane complex; 2-hydroxy-N-[(2S)-1-hydroxy-3-methyl-1,1-diphenylbutan-2-yl]benzamide; In tetrahydrofuran; at 65℃;Inert atmosphere; |
General procedure: A 25mL two-necked flask was charged with β-hydroxyamide 1-7 (0.05mmol, 10%) in dry and fresh THF (3mL), equipped with a magnetic stirrer and a connection to the combined nitrogen/vacuum line and closed with a septum. The air in the flask was replaced by nitrogen. β-hydroxyamides 1-7 were dissolved in THF (3mL) with stirring and a solution of BH3·SMe2 (0.5mmol, 10M) complex was added at 0C by a syringe. After the mixture was stirred for 1h at 65C, the freshly distilled ketone (0.5mmol) in freshly dried THF (2mL) was added over a period of 1.5h by a syringe at the same temperature. The reaction mixture was kept stirring at the 65C until the ketone was completely consumed. After stirring for a further 30min at rt, the reaction mixture was quenched by the addition of MeOH (2mL) and extracted with CH2Cl2 three times. The combined organic extracts were washed with brine and dried over MgSO4. After evaporating the solvent under reduced pressure, the product was purified by column chromatography on silica gel using petroleum ether/EtOAc (5:1; for 4′-nitroacetophenone: 5/3) as the eluent. The ee value was determined by HPLC with Chiralcel AS-3 or Chiralcel OD-H columns. |
|
With D-(+)-glucose; In ethanol; water; at 30℃; for 24h; |
General procedure: K. marxianus, Hansenula sp., G. candidum, Candida sp., R. rubra, R. minuta, and filamentous fungi, A. niger, Trichoderma harzianum, and M. ramannianus, belong to the collection of the ‘Departamento de Engenharia Bioqumica, Escola de Qumica, UFRJ’. Cells were allowed to grow for 48h at 30C under a shaking speed of 150rpm in the orbital shaker in a medium containing 1% glucose, 0.5% yeast extract, 0.5% peptone, 0.1% (NH4)2SO4, and 0.1% MgSO4·7H2O. After that period, they were harvested by centrifugation, re-suspended in distilled water and used for the reaction. After centrifugation, the cells (12g/L, dried weight) were added to the 250mL Erlenmeyer containing: 5% glucose in a final volume of 50mL distilled water. After 30min of addition of the microorganisms, the substrate (previously dissolved in 1mL of ethanol) was added to the 50mL of the mixture to give a final solution with substrate concentration of 7.2mM or 14.4mM. The reaction was carried out for 24h at 30C under a shaking speed of 150rpm in the orbital shaker. After 24h, the mixture was centrifuged to separate the cells and the liquid phase was extracted with ethyl acetate. The organic phase was dried (anhydrous Na2SO4), filtered, and concentrated under vacuum. Products were analyzed by (chiral) gas chromatography (GC). |
|
With D-(+)-glucose; In ethanol; water; at 30℃; for 24h; |
General procedure: After the growing process described above, the cells (12g/L, dry weight) were centrifuged and the precipitate was re-suspended in 3mL of distilled water to obtain a cell-suspension. Then, a 1.5% w/v sodium alginate aqueous solution in distilled water (final volume of 20mL) was added to the cell-suspension, and the mixture (cell-suspension and sodium alginate aqueous solution) was dropped into a CaCl2 aqueous solution (0.1M), forming calcium alginate spheres. Spheres were filtered and washed with distilled water. After that, the spheres were added to the 250mL Erlenmeyer containing 50mL of a 5% glucose solution with substrate concentration of 7.2 or 14.4mM (substrates were previously dissolved in 1mL of ethanol before they were added to the 5% glucose solution). The reaction was carried out for 24h at 30C under a shaking speed of 150rpm in the orbital shaker. After 24h, the mixture was filtered to separate the cells and the liquid phase was extracted with ethyl acetate. The organic phase was dried (anhydrous Na2SO4), filtered, and concentrated under vacuum. Products were analyzed by (chiral) gas chromatography (GC). |
|
With Pt/Al2O3; hydrogen; (S)-2-(dibiphenyl-4-ylfluoromethyl)pyrrolidine; In toluene; at 20℃; under 750.075 Torr; for 2h;Sealed tube;Catalytic behavior; |
General procedure: Hydrogenations of ketones at atmospheric H2-pressure wereperformed in a 50-mL three-necked glass reactor at RT and atatmospheric pressure under constant flow of molecular H2 with a volumetric flow rate of 10 mL min-1 (semi-batch hydrogenation). The third opening of the reactor was sealed with a septum which allowed for addition/removal of solutions containing modifier and ketone by the use of a syringe. The stirring rate was set to 500 rpm. Hydrogenations performed at 10 bar H2-pressure were carried out in a 60-mL Hastelloy steel jacketed-reactor connected to a multi-position valve (VICI) which allows for connecting the reactor to the hydrogen and nitrogen reservoirs, and to open it to the atmosphere.The H2-pressure was controlled with a constant pressure regulator (Brooks 5688 Series). The standard reaction temperature (298 K) in the jacket was controlled with a Haake Phoenix (Thermo) water bath. The stirring rate was set to 750 rpm. The general reaction procedure for all hydrogenations was the following: the pre-reduced catalyst (50 mg Pt/Al2O3) was transferred to the reactor und reduced again in situ in 5 ml solvent under constant H2flow for 1 h. Then, the reaction was initiated by addition of modifier and ketone premixed in 5 mL solvent. The conversion and enantioselectivity in the hydrogenation were determined by gas chromatography (GC), using an Agilent Technologies 7890A gas chromatograph equipped with a flame ionisation detector (FID). Samples were injected with a split ratio of20: 1 at an injector temperature of 250C. For GC separation, a chiral capillary column (CP-Chirasil-Dex CB, 25 m length, 0.25 mm internal diameter, 0.25 m film thickness) was used. For the analysis of KPL hydrogenations, the temperature programme started at 80C, increased to 140C at 10C min-1, increased to 180C at 20C min-1, and then held for 2 min. For the analysis of MBF hydrogenations, the temperature programme started at 120C,increased to 180C at 20C min-1, and then held for 2 min. For the analysis of TFAP hydrogenations, the temperature programmestarted at 120C, held for 1 min, increased to 130C at 1C min-1, held for 1 min, increased to 140C at 10C min-1, held for 1 min,increased to 150C at 1C min-1, held for 1 min, and then increased to 180C at 40C min-1. The FID was operated at 300C with con-stant flows of hydrogen as fuel gas (30 mL min-1) and air as oxidant(400 mL min-1). Nitrogen was used as a make-up gas (25 mL min-1)and helium as a carrier gas (constant flow: 1.623 mL min-1). The target analytes could be separated: KPL (retention time 5.84 min,elution temperature 138.4C, (S)-PL (7.38 min, 167.6C), and (R)-PL (7.51 min, 170.2C); MBF (retention time 6.09 min, elutiontemperature 145.5C, (R)-MM (7.38 min, 155.2C), and (S)-MM(7.51, 155.7C); TFAP (retention time 1.75 min, elution temperature 120.8C, (S)-PTFE (10.7 min, 130.0C), and (R)-PTFE (11.1 min,130.1C). Products were identified using enantiopure alcohol products. |
|
With benzyl[(1R,2R)-2-[(diphenylphosphoroso)amino]-1,2-diphenylethyl]amine; diethylzinc; In hexane; at -20℃; for 48h;Inert atmosphere;Catalytic behavior; |
General procedure: To a solution of chiral phosphinamide chiral ligand (0.3 mmol) in toluene (1.3 mL), Et2Zn (1.3 mL, 1 M in n-hexane, 1.3 mmol) was added and the resulting mixture was stirred for 10 min at 0 C under an atmosphere of nitrogen. Next, the reaction mixture was cooled to 20 C, and α-trifluoromethyl ketone (1.0 mmol) was added dropwise. After being stirred for 48 h, the reaction was quenched by saturated NH4Cl solution (15 mL) and the mixture was extracted with EtOAc (15 mL 3). The combined organic layer was dried over MgSO4, filtered, and concentrated in vacuo to give a residue, which was purified by flash column chromatography on silica gel (hexane/EtOAc = 10:1) to give the corresponding product. |
|
With bis(1,5-cyclooctadiene)iridium(I) tetrafluoroborate; formic acid; sodium formate; (1R,2R)-N1,N-di(naphthalen-1-yl)cyclohexane-1,2-diamine; In methanol; water; at 70℃; for 22h;Inert atmosphere;Catalytic behavior; |
General procedure: In a pressure tube, 0.5 mol% of metal precursor [C16H24BF4Ir](2.48 mg, 0.005 mol) and 1 mol% of chiral amine ligand (3.66 mg,0.01 mmol) were dissolved in 2 mL of water and methanol (ratio1:1) and stirred at room temperature for 1 h under argon atmo-sphere. Then formic acid (2.5eq, 0.1 mL), sodium formate (2.5eq,170 mg) and 1eq of ketone substrate (1 mmol) were introduced.The reaction mixture was stirred at 500 rpm and heated at 70C for22 h. After that, the tube was cooled to room temperature; and theorganic compound was extracted with either with ethyl acetate orCH2Cl2, then the solution was dried over Na2SO4, filtrated and con-centrated under reduced pressure. The crude material was purifiedby flash column chromatography on silica gel using cyclohex-ane/ethyl acetate as gradient eluent (90:10-7:3). After evaporation,alcohols were obtained as oil or solid. The products were identifiedby NMR. The conversion and the enantioselectivity were deter-mined by chiral GC or chiral HPLC analysis (Scheme 1). |
|
With dimethylsulfide borane complex; (1R,2S,3R,5R)-2-(1',3',2'-dioxaborolan-2'-yloxy)apopinan-3-amine; In tetrahydrofuran; at 20℃; for 1h; |
General procedure: To a solution of 1 (0.005-0.01 mmol, 0.5-1 mol %) in dry THF(3 mL) at room temperature, a solution of BH3SMe2 (10 M,100 lL, 1 mmol) in THF (2 mL) was added dropwise at a rate of3.2 mL per hour using a syringe pump. At the same time a solutionof ketone (1 mmol) in THF (2 mL) was also added to the reactionflask at a rate of 3 mL per hour. After the addition of both reagents,the reaction mixture was stirred for 20 min, quenched by the additionof MeOH (1 mL) at room temperature, and stirred for 30 min. Subsequently, the solvents were evaporated under vacuum and theproduct was isolated by column chromatography using hexane/EtOAc (4:1) as the eluent. |
|
With dimethylsulfide borane complex; 3-hydroxy-N-[(1R,2S)-cis-2-hydroxy-2,3-dihydro-1H-inden-1-yl]naphthalene-2-carboxamide; In tetrahydrofuran; at 65℃;Inert atmosphere;Catalytic behavior; |
General procedure: A 25mL two-necked flask was charged with β-hydroxyamide 1-7 (0.05mmol, 10%) in dry and freshly distilled THF (3mL), equipped with a magnetic stirrer and a connection to a combined nitrogen/vacuum line and closed with a septum. The air in the flask was replaced by nitrogen. β-Hydroxyamides 1-7 were dissolved in THF (3mL) under stirring after which a solution of BH3.SMe2 (0.5mmol, 10M) complex was added at 0C by a syringe. After the mixture was stirred for 1h at 65C, the freshly distilled ketone (0.5mmol) in dry and freshly distilled THF (2mL) was added over a period of 1.5h by syringe at the same temperature. The reaction mixture was stirred at 65C until the ketone was completely consumed. After stirring for a further 30min at rt, the reaction mixture was quenched by the addition of MeOH (2mL) and extracted with CH2Cl2 (3×3mL). The combined organic extracts were washed with brine and dried over MgSO4. After evaporating the solvent under reduced pressure, the product was purified by column chromatography on silica gel using petroleum ether/EtOAc (5:1; for 4′-nitroacetophenone: 5/3) as eluent. The ee value was determined by HPLC with Chiralcel AS-3 or Chiralcel OD-H columns |
|
With dimethylsulfide borane complex; 3-hydroxy-N-[(1S)-2-hydroxy-1-phenylethyl]naphthalene-2-carboxamide; In tetrahydrofuran; at 65℃;Inert atmosphere;Catalytic behavior; |
General procedure: A 25mL two-necked flask was charged with β-hydroxyamide 1-7 (0.05mmol, 10%) in dry and freshly distilled THF (3mL), equipped with a magnetic stirrer and a connection to a combined nitrogen/vacuum line and closed with a septum. The air in the flask was replaced by nitrogen. β-Hydroxyamides 1-7 were dissolved in THF (3mL) under stirring after which a solution of BH3.SMe2 (0.5mmol, 10M) complex was added at 0C by a syringe. After the mixture was stirred for 1h at 65C, the freshly distilled ketone (0.5mmol) in dry and freshly distilled THF (2mL) was added over a period of 1.5h by syringe at the same temperature. The reaction mixture was stirred at 65C until the ketone was completely consumed. After stirring for a further 30min at rt, the reaction mixture was quenched by the addition of MeOH (2mL) and extracted with CH2Cl2 (3×3mL). The combined organic extracts were washed with brine and dried over MgSO4. After evaporating the solvent under reduced pressure, the product was purified by column chromatography on silica gel using petroleum ether/EtOAc (5:1; for 4′-nitroacetophenone: 5/3) as eluent. The ee value was determined by HPLC with Chiralcel AS-3 or Chiralcel OD-H columns |
|
With tris(triphenylphosphine)ruthenium(II) chloride; (3aR,5R,7R,7aS)-2-(2-(diphenylphosphino)phenyl)-6,6-dimethyl-3a,4,5,6,7,7a-hexahydro-5,7-methanobenzo[d]oxazole; potassium tert-butylate; isopropyl alcohol; for 0.5h;Inert atmosphere; Reflux; |
General procedure: In a two-neck flask (25 mL), under an inert atmosphere, wereplaced a catalyst solution (0.05 M in isopropanol, 100 lL, 5 lmol),a degassed 0.125 M solution of sodium hydroxide or potassium tert-butoxide in isopropanol (1 mL, 0.125 mmol), and degassed isopropanol(4 mL). After stirring for 15 min. at room temperature acetophenone (120 mg, 1 mmol) was added and the reaction mixture was refluxed for 30 min. Isopropanol was removed on rotary evaporator and 1-phenylethanol was isolated by flash column chromatography on silica gel (eluent: n-hexane:ethyl acetate80:20). |
|
With trimethylamine-N-oxide; C27H18FeO4; hydrogen; In isopropyl alcohol; at 80℃; under 37503.8 Torr; for 22h;Autoclave; |
General procedure: Under argon atmosphere, the pre-catalyst was dispensed as DCM solution into oven-dried glass tubes fitted in an aluminum block inside a Schlenk tube. After removing DCM under vacuum, a 0.2m solution of Me3NO in iPrOH (0.1mL, 0.002mmol) was dispensed. The resulting mixture was stirred at room temperature for 20min, during which a deep orange color gradually developed. iPrOH (0.3mL) and the substrate (0.2mmol) were added in each vial. Each vial was capped with a Teflon septum pierced by a needle, the block was transferred into the autoclave, and stirring was started. After purging four times with hydrogen, the reaction was pressurized at 50bar and heating was started (80C). The reactions were stirred for 22h under hydrogen pressure at 80C. After cooling down to room temperature, the mixtures were filtered through a short part of celite and then analyzed for conversion and e.e. determination. When needed, product purification was carried out by flash column chromatography (hexane/AcOEt eluent mixtures). |
|
With trimethylamine-N-oxide; C19H26FeO4Si; hydrogen; In isopropyl alcohol; at 80℃; under 37503.8 Torr; for 22h;Autoclave; |
General procedure: Under argon atmosphere, the pre-catalyst was dispensed as DCM solution into oven-dried glass tubes fitted in an aluminum block inside a Schlenk tube. After removing DCM under vacuum, a 0.2m solution of Me3NO in iPrOH (0.1mL, 0.002mmol) was dispensed. The resulting mixture was stirred at room temperature for 20min, during which a deep orange color gradually developed. iPrOH (0.3mL) and the substrate (0.2mmol) were added in each vial. Each vial was capped with a Teflon septum pierced by a needle, the block was transferred into the autoclave, and stirring was started. After purging four times with hydrogen, the reaction was pressurized at 50bar and heating was started (80C). The reactions were stirred for 22h under hydrogen pressure at 80C. After cooling down to room temperature, the mixtures were filtered through a short part of celite and then analyzed for conversion and e.e. determination. When needed, product purification was carried out by flash column chromatography (hexane/AcOEt eluent mixtures). |
|
With potassium tert-butylate; [((1S,2S)-1-((4R,11bS)-3H-dinaphtho[2,1-c:1',2'-e]phosphepin-4(5H)-yl)-1-phenyl-2-propanamine)Mn(CO)3Br]; isopropyl alcohol; at 50℃; for 14h;Schlenk technique; |
General procedure: To a solution of ketone (2 mmol) in 2-propanol (19 mL) were added in this order a stock solution of complex 1 in 2-propanol (0.5 mL, 0.02 mol.L-1), and a stock solution of tBuOK in 2-propanol (0.5 mL, 0.04 mol.L-1) at 30 C. The mixture was stirred for 14 h in an oil bath at 30 C. The solution was then filtered through a small pad of silica (4 cm high in a column with a diameter of about 1 cm). The silica was washed with ethyl acetate, volatiles were removed under reduced pressure and the conversion was determined by 1H NMR. The crude reaction mixture was purified by column chromatography (SiO2, a mixture of petroleum ether/ ethyl acetate as eluent) to afford the corresponding alcohol. This latter was analyzed by chiral GC. Enantiomeric excesses were determined by GC analyses performed on a Shimadzu GC-2010 apparatus equipped with a Supelco betaDEX 120 column (30m0.25 mm) using Helium as the vector gas. |
|
With formic acid; C18H23ClN2O2RuS; triethylamine; at 40℃; for 24h; |
General procedure: A solution of the ruthenium complex (0.015 mmol) in anazeotropic mixture of formic acid/triethylamine (5:2) (1.5 ml) was stirred for 30 min at 40C. The ketone substrate (3.0 mmol) was then added and the reaction was stirred at 40C for 24 h. The reaction was then diluted with DCM (20 ml) and the organic solution washed with sat. NaHCO3(aq) (3x 15 ml). The organic phase was dried over Na2SO4, filtered the dried under reduced pressure.Residual metal-containing residues were removed from the resulting oil by purification through a plug of silica in a pipette eluting with a solvent system of EtOAc/Hexane (1:1). Solvent was removed under reduced pressure then the sample was analysed by chiral GC. GC chiral column: Restek RT-βDEXsm, 30 m x 0.25 mm x 0.25 μm using the methods described earlier. |






 Chemistry
Chemistry
 Pharmaceutical Intermediates
Pharmaceutical Intermediates
 Inhibitors/Agonists
Inhibitors/Agonists
 Material Science
Material Science






 HazMat Fee +
HazMat Fee +

 For Research Only
For Research Only
 120K+ Compounds
120K+ Compounds
 Competitive Price
Competitive Price
 1-2 Day Shipping
1-2 Day Shipping