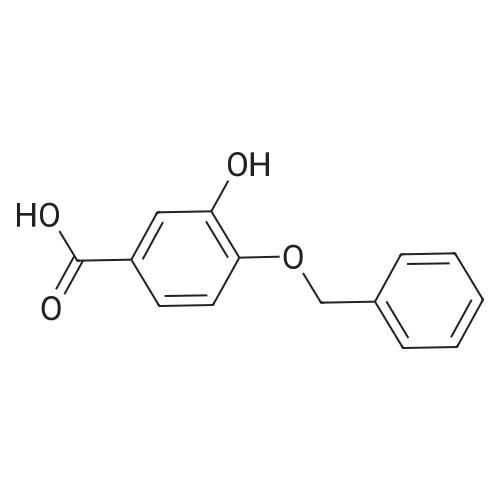
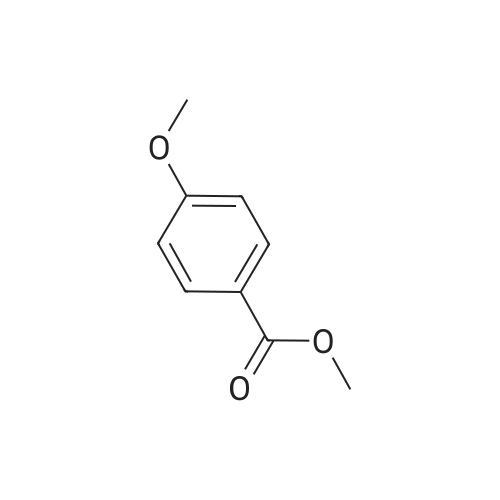
| 100% |
With potassium carbonate; In N,N-dimethyl-formamide; at 100℃; for 3.0h; |
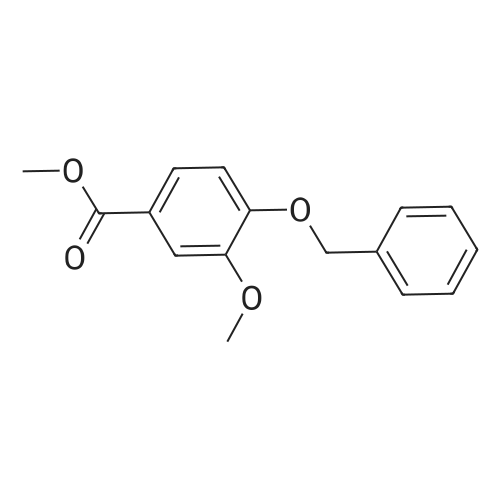
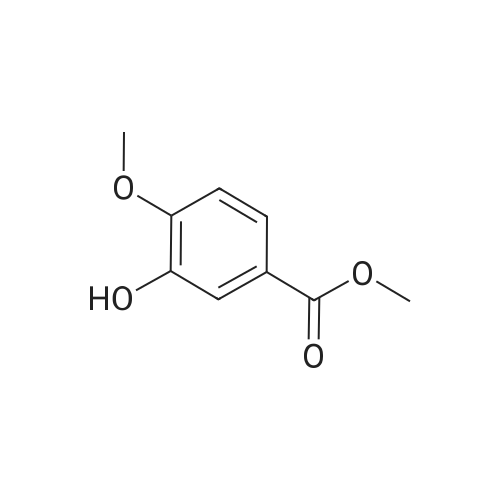
Step 1. To a solution of methyl vanillate or methyl isovanillate (7.29 g, 40 mmol) in dimethylformamide (25 mL), potassium carbonate (8.29 g, 60 mmol) and benzyl bromide (5.26 mL, 44 mmol) were added. The mixture was heated to 100 C. for 3 h. After cooling to r. t., water was added and the product was extracted several times with ethyl acetate. The combined organic phases were washed with water and brine. After drying over Na2SO4, the solvent was removed to yield methyl 4-benzyloxy-3-methoxybenzoate or methyl 3-benzyloxy-4-methoxybenzoate, respectively, quantitatively, which was used without further purification. |
| 99% |
With potassium carbonate; In N,N-dimethyl-formamide; at 100℃; for 3.0h; |
Step 1. To a solution of methyl vanillate (7.29 g, 40 mmol) in dimethylformamide (25 mL), potassium carbonate (8.29 g, 60 mmol) and benzyl bromide (5.26 mL, 44 mmol) were added. The mixture was heated to 100C for 3 h. After cooling to r.t., water was added and the product was extracted several times with ethyl acetate. The combined organic phases were washed with water and brine. After drying over Na2SO4, the solvent was removed to yield methyl 4-benzyloxy-3-methoxybenzoate (10.8 g, 39.7 mmol, 99 %) as a grey solid which was used without further purification. LC/ESI-MS: m/z = 273 [M+H]+; Rt = 3.82 min. |
| 99% |
With potassium carbonate; In N,N-dimethyl-formamide; at 100℃; for 3.0h; |
Step 1. To a solution of methyl vanillate (7.29 g, 40 mmol) in dimethylformamide (25 mL), potassium carbonate (8.29 g, 60 mmol) and benzyl bromide (5.26 mL, 44 mmol) were added. The mixture was heated to 100 C. for 3 h. After cooling to r.t., water was added and the product was extracted several times with ethyl acetate. The combined organic phases were washed with water and brine. After drying over Na2SO4, the solvent was removed to yield methyl 4-benzyloxy-3-methoxybenzoate (10.8 g, 39.7 mmol, 99%) as a grey solid which was used without further purification. LC/ESI-MS: m/z=273 [M+H]+; Rt=3.82 min. |
| 98% |
With potassium carbonate; In N,N-dimethyl-formamide; at 100℃; for 6.0h;Inert atmosphere; |
[0506] To a solution of compound Int-1-1 (54.2 g, 0.30 mol) in DMF (200 mL) was added K2CO3 (61.6 g, 0.45 mol) and benzyl bromide (39.0 mL, 0.33 mol) under N2 atmosphere. After stirring for 6 h at 100C., the mixture was cooled to room temperature and diluted with distilled water (100 mL) and EA (200 mL2). The organic layer was dried over anhydrous Na2SO4, filtered and concentrated under reduced pressure. The residue was purified by column chromatography to obtain compound Int-1-2 (79.8 g, 98%). [0507] 1H NMR (400 MHz, CDCl3) delta 7.60 (dd, J=6.4, 2.0 Hz, 1H), 7.56 (d, J=2.0 Hz, 1H), 7.44-7.31 (m, 5H), 6.89 (d, J=8.4 Hz, 1H), 5.22 (s, 2H), 3.94 (s, 3H), 3.88 (s, 3H). |
| 97% |
With potassium carbonate; In N,N-dimethyl-formamide; at 60℃; for 5.0h; |
4.1.3 Methyl 4-benzyloxy-3-methoxybenzoate (11) To a solution of methyl vanillate (10) (1.82 g, 10 mmol) in DMF (10 mL) were added K2CO3 (1.38 g, 10 mmol) and benzyl bromide (2.05 g, 12 mmol). The mixture was heated at 60 C for 5 h. After cooling to room temperature, water was added and the product was extracted with CH2Cl2. The organic phase was washed with water and brine. After drying over Na2SO4, the solvent was removed to yield intermediate 11 as a white solid. Yield: 97%, mp: 77-79 C. |
| 95% |
With potassium carbonate; In N,N-dimethyl-formamide; at 60℃; for 2.0h; |
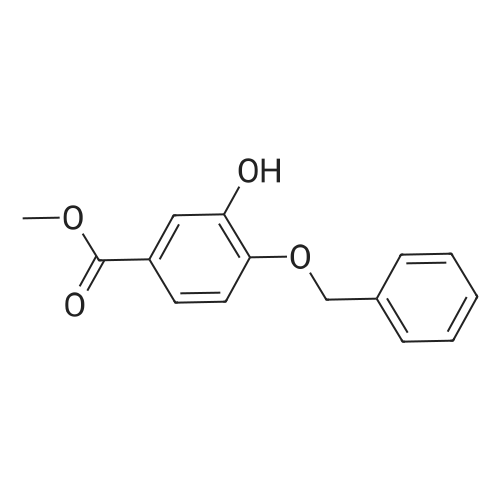
Step 2a. Methyl 4-(benzyloxy)-3-methoxybenzoate (Compound 202) To a mixture of compound 201 (18.2 g, 0.1 mol), potassium carbonate (34.55 g, 0.25 mol) in N,N-dimethylformamide was added benzylbromide (14.5 ml, 0.105 mol) dropwise. The reaction was then heated to 60 C. and stirred for 2 hours. The mixture was cooled to room temperature and was filtered. The filtrate was concentrated and the residue was dissolved in ethyl acetate 500 mL. The organic layer was washed with water and brine (100 mL), dried over MgSO4, filtered and concentrated to give the title compound 202 as a white solid (26 g, 95%): LCMS: 273 [M+1]+. |
| 95% |
With potassium carbonate; In N,N-dimethyl-formamide; at 80℃; for 6.0h; |
A 250 mL, round-bottomed flask with a stirring bar, a solution of 4-hydroxy-3-methoxybenzoic acid (20 g, 118.94 mmol) was added slowly to a solution of methanol (100 mL) and concentrated sulfuric acid (10 mL). After being stirred for 12 h at reflux, saturated solution of sodium bicarbonate was added to adjust the pH to 7. Dichloromethane was added and the mixture was then filtered and the organic phase evaporated on a rotary evaporator and to obtain the compound 2 (20.37 g, 94 %). Compound 2 (20.4 g, 111.98 mmol) was added into a 500 mL, round-bottomed flask with a stirring bar, then benzyl bromide (18 mL), potassium carbonate (22 g, 156.8 mmol), DMF (200 mL) were added. It was stirred for 6 h at 80 C. Then the reaction system was poured into right amount of water, white solid (3) was obtained by filtration (28.97 g, 95 %). Compound 3 (16.54 g, 60.74 mmol) was dissolved in CH3COOH (50 mL) and then added into a 250 mL, round bottomed flask with a stirring bar. Then HNO3 (25 mL) was added into the system slowly to keep the temperature of the reaction above 5 C. The reaction temperature was raised to 50 C and kept for another 2 h. After that the system was poured into water and pale yellow solid (I) was obtained6-8 (18.3 g, 95%, m.p.: 134-135 C). |
| 91% |
With potassium carbonate; In acetone; at 45℃; for 3.5h; |
To a mechanically stirred solution of methyl vanillate (103.5 g, 0.568 mol) and benzyl bromide (101.36 ml, 0.852 mol) in acetone (800 ml) at room temperature powdered K2CO3 (196.25 g, 1.4 mol) is added The reaction is heated to 45 C for 3.5 hours, cooled and filtered. The filtrate is concentrated in vacuo and the residue dissolved in EtOAc (300 ml) and washed with water (100 ml) three times saturated. NaHCtheta3 (100 ml x 2), and brine. The organic layer is dried over Na2SO4 and concentrated in vacuo to provide 224.72g of a white solid. The solid is then triturated in hexane (300 ml) and filtered to provide methyl 4-(benzyloxy)-3-methoxybenzoate (141.45 g, 91%). |
| 86.6% |
With potassium carbonate; In acetone; for 12.0h;Heating / reflux; |
D) 4-Benzyloxy-3-methoxy benzoic acid methyl ester; Potassium carbonate (3.45 g; 25 mmol) was added to a solution of 4-hydroxy- 3-methoxy benzoic acid methyl ester (3.6 g; 20 mmol) and benzyl bromide (3.42 g; 20 mmol) in acetone (100 ml). The reaction mixture was refluxed for 12 hrs. After the removal of the solvent under reduced pressure, the residue was partitioned between ethyl acetate (150 ml) and water (50 ml). The ethyl acetate layer was washed with water (50 ml) and dried over anhydrous magnesium sulfate. Removal of the solvent under reduced pressure provided 4.64 g of 4-benzyloxy-3-methoxy benzoic acid methyl ester (Yield = 86.6%) |
| 82.2% |
With potassium carbonate; In N,N-dimethyl-formamide; at 100℃; for 3.0h; |
A solution of methyl 4-hydroxy-3-methoxybenzoate 38 (3.0 g, 16.5 mmol) in DMF (100 mL) was added benzyl bromide (4.22 g, 24.7 mmol) and K2CO3 (4.56 g, 32.9 mmol). The reaction mixture was stirred at 100 C. for 3 h. The mixture was concentrated in vacuo and was dissolved in water (50 mL), extracted with EtOAC (30 mL*2), washed with NaCl (30 mL), dried over Na2SO4. It was concentrated and purified by silica chromatography (0-30% EtOAc in petroleum ether) to give methyl 4-(benzyloxy)-3-methoxybenzoate 39 (3.8 g, 13.5 mmol, 82.2% yield) as a white solid. LCMS (5-95AB/1.5 min): RT=0.787 min, [M+H]+ 272.9 |
|
With potassium carbonate; In acetone; for 4.0h;Heating / reflux; |
(1) Benzyl bromide (3.59 ml) and potassium carbonate (5.69 g) were added to an acetone (50 ml) solution of methyl 4-hydroxy-3-methoxybenzoate (5.00 g), and heated at reflux for 4 hours. After removing solid matter by filtration and concentrating the filtrate under reduced pressure, the residue was dissolved in ethyl acetate, and washed with saturated aqueous sodium hydrogencarbonate solution and saturated brine. The organic layer was dried over MgSO4, and the solvent was evaporated under reduced pressure, thereby giving 7.47 g of methyl 4-benzyloxy-3-methoxybenzoate.(2) An aqueous 2 M sodium hydroxide solution (13.72 ml) was added to an ethanol (15 ml) solution of the above-obtained 4-benzyloxy compound (7.47 g), and heated at reflux for 2 hours. After evaporating the solvent under reduced pressure, 2M hydrochloric acid was added, and the thus-precipitated crystals were filtered, thereby giving 7.00 g of the desired compound. |
|
With potassium carbonate; In N,N-dimethyl-formamide; at 100℃; for 3.0h; |
To a solution of methyl vanillate or methyl isovanillate (7.29 g, 40 mmol) in dimethylformamide (25 mL), potassium carbonate (8.29 g, 60 mmol) and benzyl bromide (5.26 mL, 44 mmol) were added. The mixture was heated to 100 C. for 3 h. After cooling to r.t., water was added and the product was extracted several times with ethyl acetate. The combined organic phases were washed with water and brine. After drying over Na2SO4, the solvent was removed to yield methyl 4-benzyloxy-3-methoxybenzoate or methyl 3-benzyloxy-4-methoxybenzoate, respectively, quantitatively, which was used without further purification. |
|
With potassium carbonate; In acetone; |
Commercially available methyl 4-hydroxy-3-methoxy-benzoate 1 was converted in 5 steps to 4-(3-fluoroanilino)-6-methoxy-7-benzyloxy-quinazolme 15 (35% overall yield).[17,18] The only significant modification involved an improved workup of the nickel chloride hexahydrate-sodium borohydride reduction of the nitro intermediate. Debenzylation with trifluoroacetic acid to give the 7-hydroxy intermediate 16, followed by alkylation with propargyl bromide in acetonitrile, gave 13 in a 65% yield for 2 steps. This method involved an additional step, and the overall yield was greater. In addition, the final 7-hydroxy intermediate 16 could be used to generate other products [19]. |
|
With potassium carbonate; In N,N-dimethyl-formamide; at 20℃; for 4.0h; |
To a solution of methyl vanillate (5.0 g, 27.4 mmol) in DMF (50 ml) was added K2CO3 (4.92 g, 35.6 mmol) and benzyl bromide (3.9 ml, 32.9 mmol). The reaction was stirred at room temperature, under nitrogen, for 4 hours, poured into water (100 ml) and stirred for 30 minutes. The aqueous layer was extracted with EtOAc and the organic washed with saturated aqueous NaHCO3, water, brine, dried (MgSO4) and concentrated. The residue was recrystallised from heptane to provide the title compound as a cream coloured solid (6.35 g). LC/MS purity: 100 %, m/z 566 [M+H]+. 1H NMR (400 MHz, CDCI3) delta: 7.60 (1H, dd, J=8.2, 8.5 Hz), 7.56 (1H, d, J=2.2 Hz), 7.45-7.41 (2H, m), 7.40-7.35 (2H, m), 7.34-7.29 (1 H, m), 6.89 (1 H, d, J=8.5 Hz), 5.22 (2H, s), 3.94 (3H, s), 3.88 (3H, s). |
|
With potassium carbonate; In N,N-dimethyl-formamide; at 20℃; for 72.0h; |
To a solution of compound 1 (500 g, 2.74 mol) in DMF (2.50 L) was added K2C03 (757.39 g, 5.48 mol) in one portion, following BnBr (702.93 g, 4.11 mol, 488.15 mL) was added to the mixture in portions, the mixture was stirred at 20 C for 72 h. TLC (petroleum ether: ethyl acetate = 5: 1) showed that the reaction was completed. The mixture was quenched by pouring into ice-water (3 L) and the precipitated solid was collected by filtration, the filter cake was triturated with petroleum (500 mL x 2). Filtered and dried under vacuum to give compound 2 (1.00 kg, crude) as a white solid which was used in the next step without any further purification. (0193) 1H NMR: (CDC13 400MHz): delta 7.62-7.59 (dd, J1= 2.0 Hz, J2 = 8.4 Hz, 1H), 7.57-7.56 (d, / = 2.0 Hz, 1H), 7.44-7.26 (m, 5H), 6.91-6.89 (d, J = 8.4 Hz, 1H), 5.21 (s, 2H), 3.94 (s, 3H), 3.88 (s, 3H). |






 Chemistry
Chemistry
 Pharmaceutical Intermediates
Pharmaceutical Intermediates
 Inhibitors/Agonists
Inhibitors/Agonists
 Material Science
Material Science







 For Research Only
For Research Only
 120K+ Compounds
120K+ Compounds
 Competitive Price
Competitive Price
 1-2 Day Shipping
1-2 Day Shipping