| 54% |
|
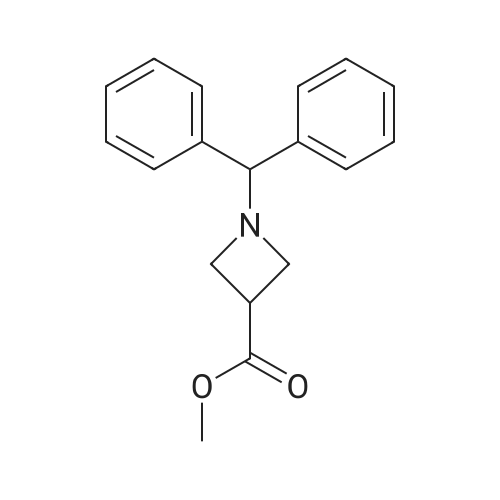
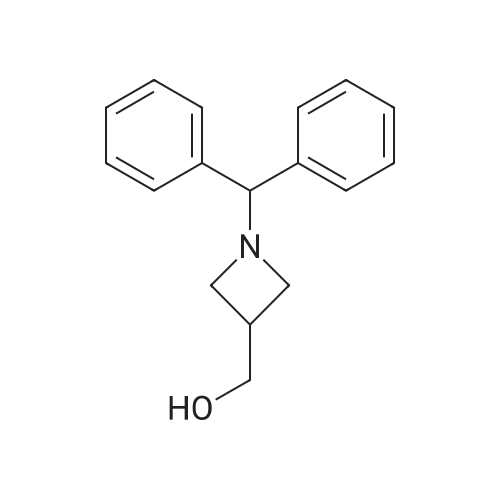
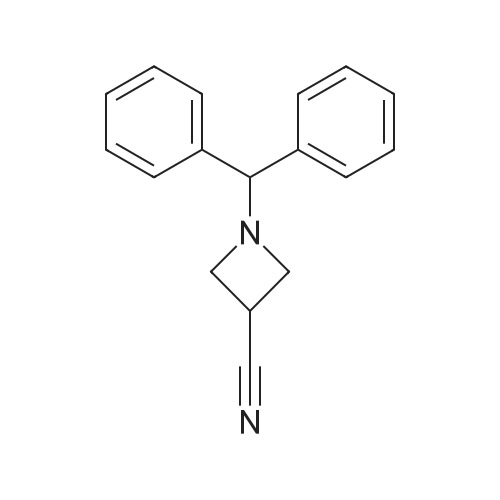
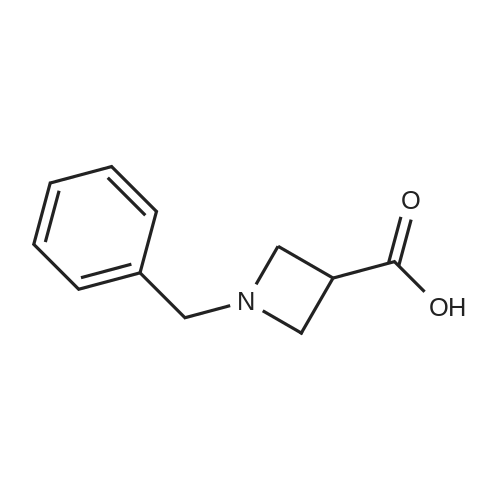
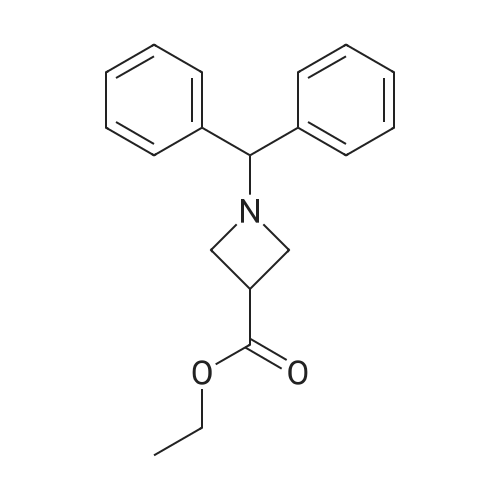
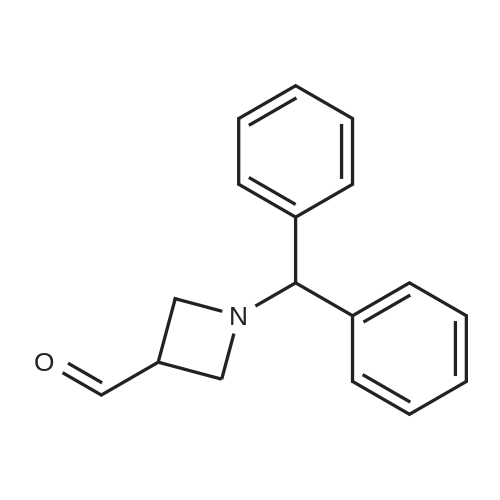
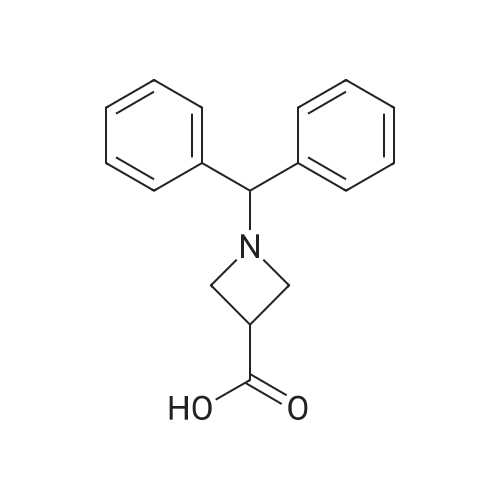
1-Benzhydryl-3-azetidinecarboxylic acid (3.12 g) was suspended in tetrahydrofuran (60 ml), and cooled in an ice-methanol bath under a nitrogen atmosphere. Triethylamine (1.96 ml) was added dropwise, and a solution of ethyl chlorocarbonate (1.34 ml) in tetrahydrofuran (5 ml) was added dropwise over 20 minutes. After the addition, the reaction mixture was stirred at the same temperature for 30 minutes. The reaction mixture was filtered, and the residue was washed with tetrahydrofuran (30 ml). The filtrate was added dropwise to a solution of an aqueous (15 ml) solution of sodium borohydride (1.33 g) cooled in an ice water bath over 15 minutes. After the addition, the reaction mixture was stirred at room temperature. 1N hydrochloric acid (35 ml) was gradually added to the reaction mixture to degrade excess sodium borohydride, and a 1N aqueous solution of sodium hydroxide (35 ml) was added. This was extracted with ethyl acetate (100 ml). The organic layer was washed with brine, and dried over anhydrous sodium sulfate. The solvent was concentrated, and the residue was dried under reduced pressure to give the title compound (1.59 g, 54%) as a pale brown solid.1H-NMR Spectrum (CDCl3) δ (ppm): 2.57 (1H, m), 3.03 (2H, m), 3.24 (2H, m), 3.80 (2H, d, J=5.2 Hz), 4.33 (1H, s), 7.15-7.45 (10H, m).ESI-MS (m/z): 254[M+H]+. |
| 54% |
|
(Reference Example F-4) 1-Benzhydryl-3-(hydroxymethyl)azetidine 1-Benzhydryl-3-azetidinecarboxylic acid (3.12 g) was suspended in tetrahydrofuran (60 ml) and cooled under a nitrogen atmosphere in an ice-ethanol bath. Triethylamine (1.96 ml) was added dropwise, and a solution of ethyl chlorocarbonate (1.34 ml) in tetrahydrofuran (5 ml) was added dropwise over 20 min. After the dropwise addition, stirring was carried out at the same temperature for 30 min. The reaction mixture was filtered and washed with tetrahydrofuran (30 ml). The filtrate was added dropwise over 15 min to an aqueous (15 ml) solution of sodium borohydride (1.33 g) cooled in an ice water bath. Upon completion of the dropwise addition, the reaction mixture was stirred at room temperature. To the reaction mixture was gradually added 1N hydrochloric acid (35 ml) to decompose excess sodium borohydride, and a 1N aqueous solution of sodium hydroxide (35 ml) was added. This was extracted with ethyl acetate (100 ml). The organic layer was washed with brine, and dried over anhydrous sodium sulfate. The solvent was concentrated, and the residue was dried under reduced pressure to provide the titled compound as a pale brown solid (1.59 g, 54 %). 1H-NMR Spectrum (CDCl3) δ (ppm): 2.57 (1H, m), 3.03 (2H, m), 3.24 (2H, m), 3.80 (2H, d, J = 5.2 Hz), 4.33 (1H, s), 7.15-7.45 (10H, m). ESI-MS (m/z):254[M+H]+. |
| 54% |
|
(Production Example 102) 1-Benzhydryl-3-(hydroxymethyl)azetidine 1-Benzhydryl-3-azetidinecarboxylic acid (3.12 g) was suspended in tetrahydrofuran (60 ml) and cooled under a nitrogen atmosphere in an ice-ethanol bath. Triethylamine (1.96 ml) was added dropwise, and a solution of ethyl chlorocarbonate (1.34 ml) in tetrahydrofuran (5 ml) was added dropwise over 20 min. After the dropwise addition, stirring was carried out at the same temperature for 30 min. The reaction mixture was filtered and washed with tetrahydrofuran (30 ml). The filtrate was added dropwise over 15 min to an aqueous (15 ml) solution of sodium borohydride (1.33 g) cooled in an ice water bath. Upon completion of the dropwise addition, the reaction mixture was stirred at room temperature. To the reaction mixture was gradually added 1N hydrochloric acid (35 ml) to decompose excess sodium borohydride, and a 1N aqueous solution of sodium hydroxide (35 ml) was added. This was extracted with ethyl acetate (100 ml). The organic layer was washed with brine, and dried over anhydrous sodium sulfate. The solvent was concentrated, and the residue was dried under reduced pressure to provide the titled compound as a pale brown solid (1.59 g, 54 %). 1H-NMR Spectrum (CDCl3) δ (ppm): 2.57 (1H, m), 3.03 (2H, m), 3.24 (2H, m), 3.80 (2H, d, J = 5.2 Hz), 4.33 (1H, s), 7.15-7.45 (10H, m). ESI-MS (m/z):254[M+H]+. |






 Chemistry
Chemistry
 Pharmaceutical Intermediates
Pharmaceutical Intermediates
 Inhibitors/Agonists
Inhibitors/Agonists
 Material Science
Material Science







 For Research Only
For Research Only
 120K+ Compounds
120K+ Compounds
 Competitive Price
Competitive Price
 1-2 Day Shipping
1-2 Day Shipping