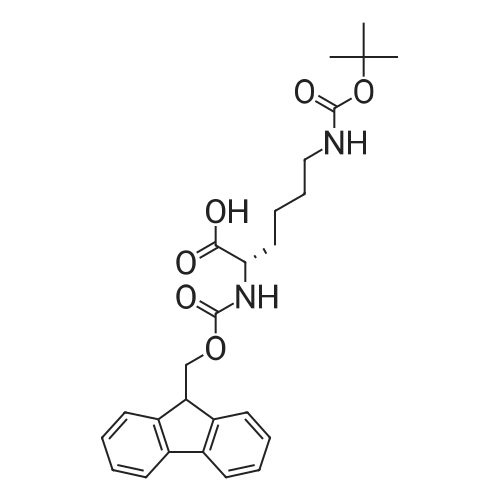
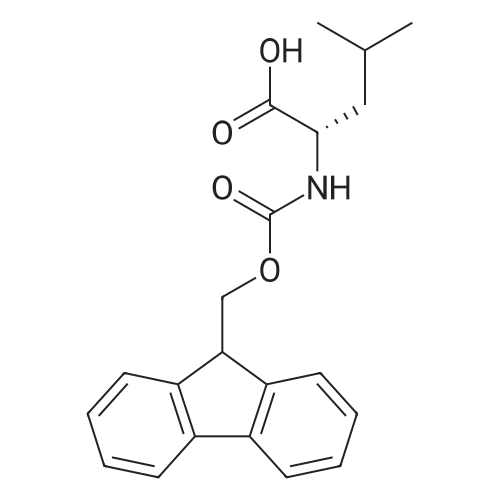
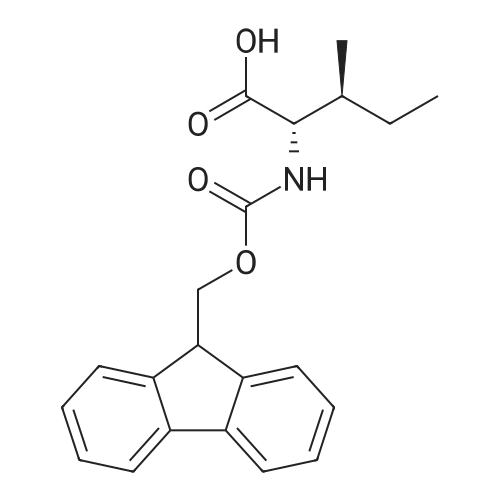
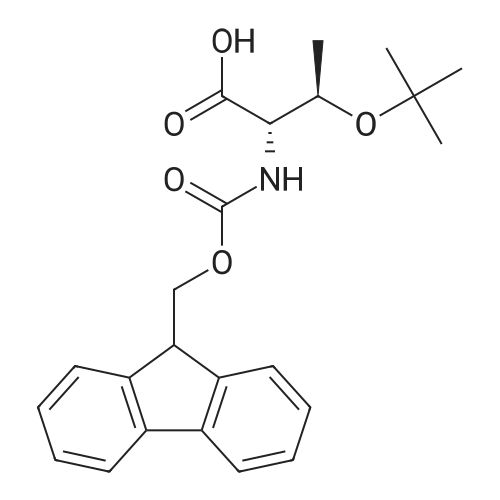
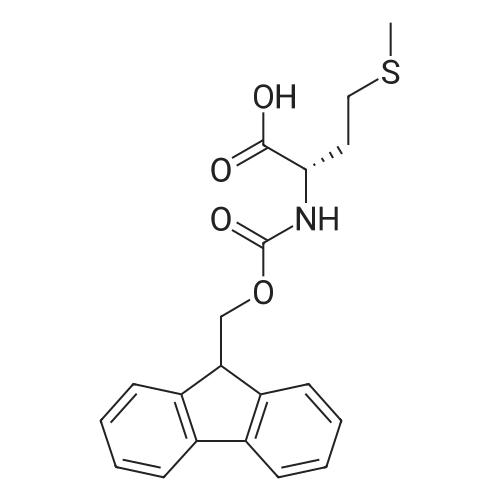
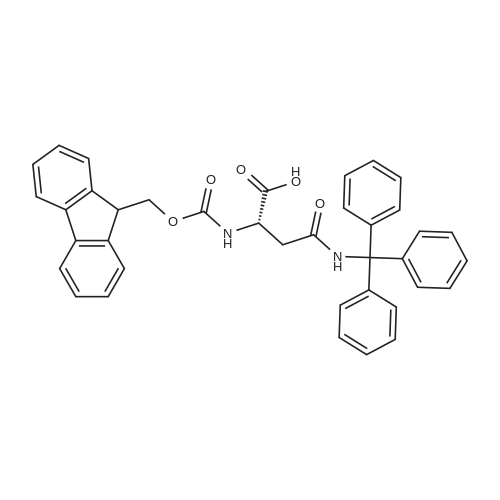
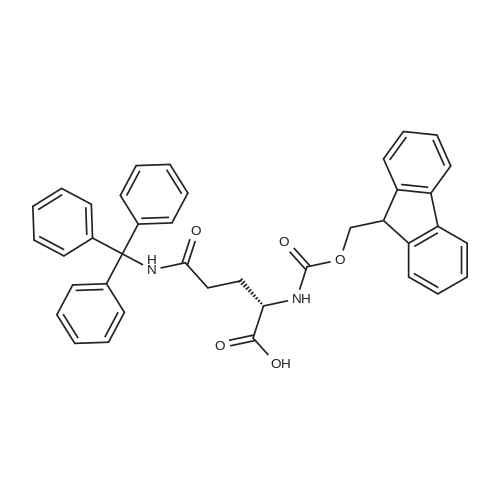
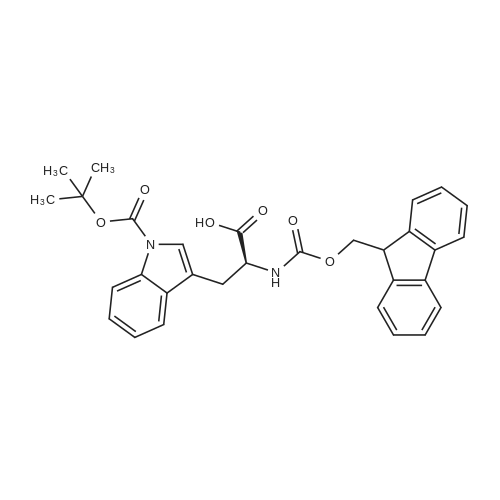
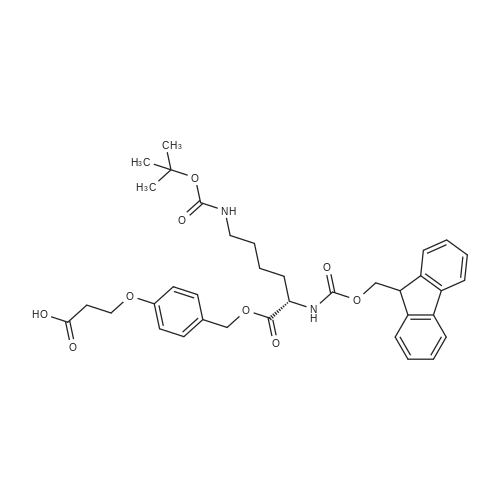
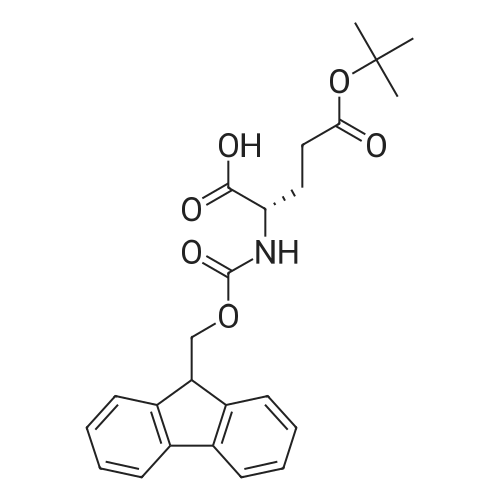
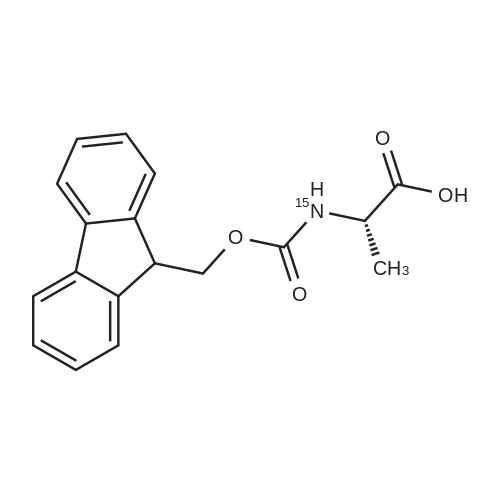
* All experimental methods are cited from the reference, please refer to the original source for details. We do not guarantee the accuracy of the content in the reference.
Lys(Boc)-Pra-Asn(Trt)-Thr(tBu)-Ala-Thr(tBu)-Ala(N3)-Ala-Pal-PEG resin (16) (0.498 g, 0.09 mmol) was cleaved with TFA/iPr3SiH/H2O (v/v/v; 95/2.5/2.5, 5.0 mL) for 2 h, peptide was isolated as described in the general section to afford 47.3 mg of crude peptide 17. The crude peptide 17 (16.3 mg) was purified by RP-HPLC on a preparative Phenomenex Gemini C18, column at a flow rate of 5 mL min-1, using a linear gradient of 1percentB to 61percentB over 60 min (ca. 1percentB per minute) and lyophilised to give the title compound 17 as a white amorphous solid (4.9 mg, 18percent).
Lys(Boc)-Pra-Asn(Trt)-Thr(tBu)-Ala-Thr(tBu)-Ala(N3)-Ala-Pal-PEG resin (16) (0.498 g, 0.09 mmol) was cleaved with TFA/iPr3SiH/H2O (v/v/v; 95/2.5/2.5, 5.0 mL) for 2 h, peptide was isolated as described in the general section to afford 47.3 mg of crude peptide 17. The crude peptide 17 (16.3 mg) was purified by RP-HPLC on a preparative Phenomenex Gemini C18, column at a flow rate of 5 mL min-1, using a linear gradient of 1percentB to 61percentB over 60 min (ca. 1percentB per minute) and lyophilised to give the title compound 17 as a white amorphous solid (4.9 mg, 18percent). Purified Lys-Pra-Asn-Thr-Ala-Thr-Ala(N3)-Ala-NH2 (17) (4.4 mg, 5.42 x 10-3 mmol) was dissolved in a mixture of water and tert-butyl alcohol (1 : 2.5, 3.5 mL in total). A stock solution of CuSO4 (0.87 mg, 5.42 x 10-3 mmol) and sodium ascorbate (2.68 mg, 13.6 x 10-3 mmol) in water (1.5 mL) was added and the mixture was microwaved for 20 min at 80 °C in a sealed reaction vessel (120 W max) to afford crude peptide (8), containing inseparable dehydroalanine by-product. The crude peptide (8) was purified by RP-HPLC on a preparative Waters XTerra.(R). Prep. C18 column at a flow rate of 10 mL/min, using a linear gradient of 1percentB to 51percentB over 50 min (ca. 1percentB per minute). Fractions were lyophilised to give the title compound 8 as a white amorphous solid (0.7 mg, 18percent), containing inseparable dehydroalanine by-product.
General procedure: Fmoc SPPS was performed on a Liberty Microwave Peptide Synthesiser (CEM Corporation, Mathews, NC) using the Fmoc/tBu strategy as previously described35 or manually starting from PAL-PEG polystyrene resin (0.21 mmol/g). For manual synthesis the following steps were undertaken: (a) Fmoc deprotection with 20percent piperidine for 5 min, then 15 min, washing with DMF 5.x.; (b) coupling of the Fmoc amino acid (5 equiv) in the presence of HBTU in DMF (4.9 equiv) and iPr2NEt (10 equiv) for 1 h and washing with DMF 5.x.. For coupling of Fmoc-Pra (1.5 equiv) and Fmoc-N3Ala (1.5 equiv), 1.45 equiv of HBTU and 4.5 equiv of iPr2NEt were used. The progress of the acylation step was monitored by the Kaiser test. A minimum amount of DMF was used for dissolution of the Fmoc amino acid. The resulting peptides were cleaved from the resin with simultaneous side chain protecting group removal by treatment with either TFA/iPr3SiH/DODT/H2O (v/v/v/v; 94/1/2.5/2.5), or with TFA/iPr3SiH/H2O (v/v/v; 95/2.5/2.5) for 2 h at room temperature. Crude peptides were precipitated and triturated with cold diethyl ether, isolated (centrifugation), dissolved in 20percent acetonitrile (aq) containing 0.1percent TFA and lyophilized. Analytical RP-HPLC was performed using a Dionex P680 (flow rate of 1 mL/min), or Dionex Ultimate U3000 system (flow rate of 0.5 mL/min or 0.2 mL/min) using Waters XTerra.(R). column (MS C18, 150 mm .x. 4.6 mm; 5 mum) or, Phenomenex Aqua column (C18, 250 mm .x. 4.6 mm; 5mu), or Phenomenex, Gemini column (C18, 50 mm .x. 2.0 mm, 5mu), using gradient systems as indicated in the Supplementary data. The solvent system used was A (0.1percent TFA in H2O) and B (0.1percent TFA in acetonitrile) with detection at 210 nm, 254 nm, and 280 nm****. The ratio of products was determined by integration of spectra recorded at 210 nm. Peptide masses were confirmed by an inline Thermo Finnegan MSQ mass spectrometer using ESI in the positive mode. When appropriate, a Bruker micrOTOF-Q II mass spectrometer was used for ESI-MS analysis (positive mode). Infrared spectra were obtained using a Perkin Elmer Spectrum One Fourier Transform infrared spectrometer with a universal ATR sampling accessory. Peptide purification was performed using a Waters 600E or Dionex Ultimate U3000 system using a Waters XTerra.(R). column (C18, 300 mm .x. 19 mm; 10 mum), or Phenomenex Gemini C18, 250 mm .x. 10 mm; 5 mum column. Gradient systems were adjusted according to the elution profiles and peak profiles obtained from the analytical RP-HPLC chromatograms. Fractions were collected, analysed by either RP-HPLC or ESI-MS, pooled and lyophilised three times from 10 mM aq HCl.
General procedure: Fmoc SPPS was performed on a Liberty Microwave Peptide Synthesiser (CEM Corporation, Mathews, NC) using the Fmoc/tBu strategy as previously described35 or manually starting from PAL-PEG polystyrene resin (0.21 mmol/g). For manual synthesis the following steps were undertaken: (a) Fmoc deprotection with 20percent piperidine for 5 min, then 15 min, washing with DMF 5.x.; (b) coupling of the Fmoc amino acid (5 equiv) in the presence of HBTU in DMF (4.9 equiv) and iPr2NEt (10 equiv) for 1 h and washing with DMF 5.x.. For coupling of Fmoc-Pra (1.5 equiv) and Fmoc-N3Ala (1.5 equiv), 1.45 equiv of HBTU and 4.5 equiv of iPr2NEt were used. The progress of the acylation step was monitored by the Kaiser test. A minimum amount of DMF was used for dissolution of the Fmoc amino acid. The resulting peptides were cleaved from the resin with simultaneous side chain protecting group removal by treatment with either TFA/iPr3SiH/DODT/H2O (v/v/v/v; 94/1/2.5/2.5), or with TFA/iPr3SiH/H2O (v/v/v; 95/2.5/2.5) for 2 h at room temperature. Crude peptides were precipitated and triturated with cold diethyl ether, isolated (centrifugation), dissolved in 20percent acetonitrile (aq) containing 0.1percent TFA and lyophilized. Analytical RP-HPLC was performed using a Dionex P680 (flow rate of 1 mL/min), or Dionex Ultimate U3000 system (flow rate of 0.5 mL/min or 0.2 mL/min) using Waters XTerra.(R). column (MS C18, 150 mm .x. 4.6 mm; 5 mum) or, Phenomenex Aqua column (C18, 250 mm .x. 4.6 mm; 5mu), or Phenomenex, Gemini column (C18, 50 mm .x. 2.0 mm, 5mu), using gradient systems as indicated in the Supplementary data. The solvent system used was A (0.1percent TFA in H2O) and B (0.1percent TFA in acetonitrile) with detection at 210 nm, 254 nm, and 280 nm****. The ratio of products was determined by integration of spectra recorded at 210 nm. Peptide masses were confirmed by an inline Thermo Finnegan MSQ mass spectrometer using ESI in the positive mode. When appropriate, a Bruker micrOTOF-Q II mass spectrometer was used for ESI-MS analysis (positive mode). Infrared spectra were obtained using a Perkin Elmer Spectrum One Fourier Transform infrared spectrometer with a universal ATR sampling accessory. Peptide purification was performed using a Waters 600E or Dionex Ultimate U3000 system using a Waters XTerra.(R). column (C18, 300 mm .x. 19 mm; 10 mum), or Phenomenex Gemini C18, 250 mm .x. 10 mm; 5 mum column. Gradient systems were adjusted according to the elution profiles and peak profiles obtained from the analytical RP-HPLC chromatograms. Fractions were collected, analysed by either RP-HPLC or ESI-MS, pooled and lyophilised three times from 10 mM aq HCl.






 Chemistry
Chemistry
 Pharmaceutical Intermediates
Pharmaceutical Intermediates
 Inhibitors/Agonists
Inhibitors/Agonists
 Material Science
Material Science







 For Research Only
For Research Only
 120K+ Compounds
120K+ Compounds
 Competitive Price
Competitive Price
 1-2 Day Shipping
1-2 Day Shipping