* All experimental methods are cited from the reference, please refer to the original source for details. We do not guarantee the accuracy of the content in the reference.
With 1,8-diazabicyclo[5.4.0]undec-7-ene In methanol for 8 h; Reflux
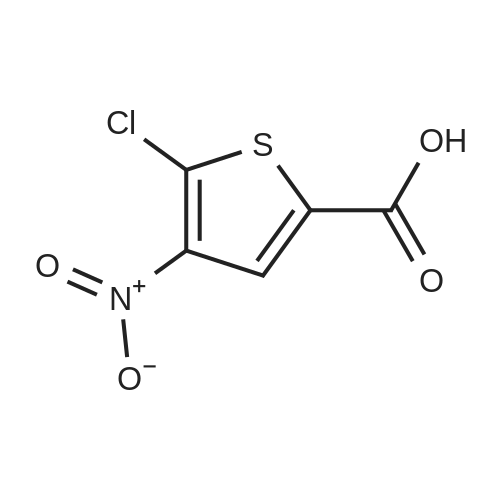
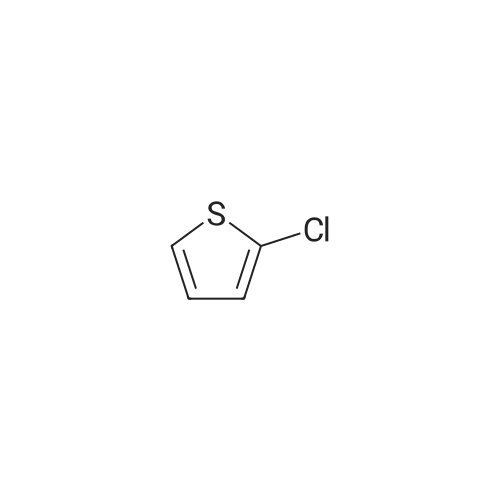
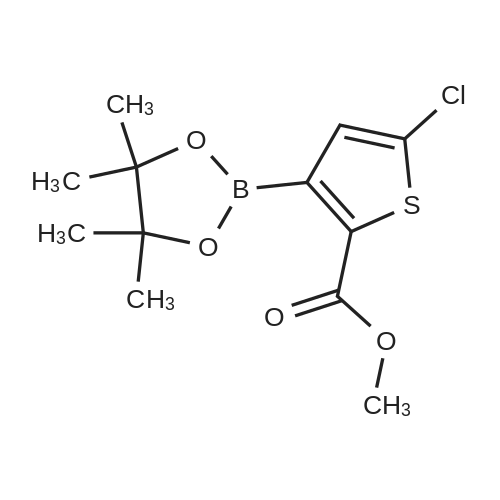
The reaction flask was prepared by adding the compound prepared as in Example 2(S) -4- [4 - [(3-amino-2-hydroxypropyl) amino] phenyl] -3-morpholinone(0.02 mol), methyl 5-chloro-2-thiophenecarboxylate (0.022 mol), DBU (0.002 mol) and 100 ml of methanol were added and the reaction was refluxed for 8 hours.The solvent was evaporated under reduced pressure and the residue was dissolved in 200 ml of dichloromethane,Followed by washing with deionized water, saturated sodium chloride solution,The organic phase was dried over anhydrous sodium sulfate and filtered, and the filtrate was evaporated under reduced pressure to remove the solvent,The residue was recrystallized from ethanol to give a white solid in a yield of 92.3percent
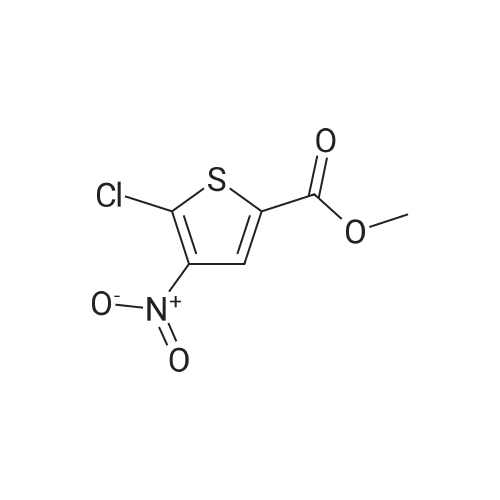
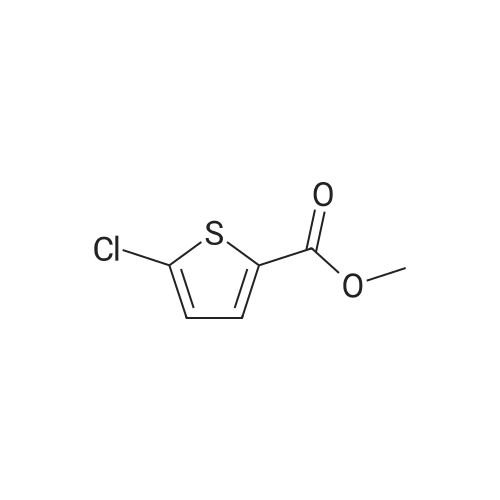
Step 2: Production of methyl 5-chloro-4-nitrothiophene-2-carboxylate Fuming nitric acid (15 ml) was cooled with ice and saturated brine, <strong>[35475-03-7]methyl 5-chlorothiophene-2-carboxylate</strong> (4.99 g, 28.3 mmol) was added by small portions, and the mixture was stirred for 1 hr. Ice water was added to the reaction mixture and the precipitated solid was collected by filtration, washed with water and vacuum dried to give methyl 5-chloro-4-nitrothiophene-2-carboxylate (3.88 g, yield 58%). 1H-NMR(400MHz, deltappm, DMSO-d6): 8.17(1H, s).
54.9%
With sulfuric acid; nitric acid; at 0℃; for 1h;
12.2 mmol (2.157 g) TEPS 63 were solved in 3.9 mL cone. H2S04, and then cooled down to 0- 10 C. A 0-5 C cold mixture of 3 mL cone. H2S04 and 1.7 mL cone. HN03 were added slowly and stirred for one hour at 0 C. Afterwards the mixture was poured on iced-water whereas a precipitate was formed. The precipitate was collected by vacuum filtration and washed three times, starting with H20, following by 5 % -sodiumhydrogencarbonat solution and again washed with H20. The product was yellow with a yield of 1.502 g (54.9 %). 1H NMR (200 MHz, chloroform-d) d 8.18 (s, 1H), d 3.94 (s, 3H). 13C NMR (50 MHz, chloroform-d) d 128.6, 53.1. MS m/z : 221 M+
47.8%
With sulfuric acid; nitric acid; at 0℃; for 3h;
Compound 16 (30.00 g, 169.86 mmol)Soluble in 60mL sulfuric acid,Nitric acid (12.84 g, 203.83 mmol)Add 12mL sulfuric acid to mix,Immediately added to the above solution,Reaction at 0C for 3 hoursThe reaction was stopped by adding 100 mL of ice water, extracted with ethyl acetate (100 mL x 3), dried over anhydrous sodium sulfate, and the solvent was recovered under reduced pressure. Column chromatography gave 18.0 g of a white solid (compound 17). The yield was 47.8%.
47.8%
With sulfuric acid; nitric acid; at 0℃; for 3h;
To a solution of compound 2 (30.0 g, 169.9 mmol, 1.0 eq) incon.H2SO4 (60.0mL) was added a mixture of con.HNO3(12.8 g, 203.8mmol, 9.2mL, 1.2 eq) in con.H2SO4 (12.0mL).The reaction was stirred at 0 C for 3 h. The reaction wasquenched with ice-water (100.0 mL), extracted with EtOAc(100.0 mL × 3). The combined organic layer was dried overNa2SO4 and concentrated under reduced pressure. The crudeproduct was purified by CombiFlash (0-10% EtOAc in PE) togive compound 3 as white solid. Yield: 47.8%; 1H NMR(400 MHz, CDCl3) : delta=8.19 (s, 1H, thienyl-H), and 3.94 (s,3H, CH3). ESI-MS: m/z=222.9[M+H]+.
With N,N,N-trimethylbenzenemethanaminium dichloroiodate; zinc(II) chloride; In acetic acid; at 70℃; for 48h;
To a solution of delta-chloro^-thiophenecarboxylic acid (5.0 g, 28.0 mmol) in acetic acid (150 mL) was added ZnCI2 (38g, 280 mmoles) and benzyltrimethylammonium dichloroiodate (20.5 g, 58.8 mmole) [Bull. Chem. Soc. <n="111"/>Jpn. 64, 2566-2568 (1991 )]. The reaction mixture was heated to 70 C for 48h and was then concentrated under vacuum. The residue was extracted with hexanes (2 x 200 ml.) and the hexane solution washed with saturated NaHCOs solution. The organic fractions were dried (Na2SO4), concentrated under vacuum and purified on silica gel (hexanes/EtOAc, 4:1 ) to give the title compound (3.8 g, 45%) as a light yellow solid: LC-MS (ES) m/z = 302 (M+H)+.
With aluminum (III) chloride; bromine; In chloroform; at 25℃; for 6h;
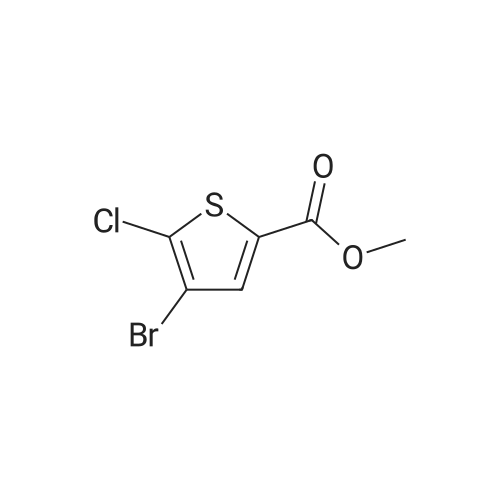
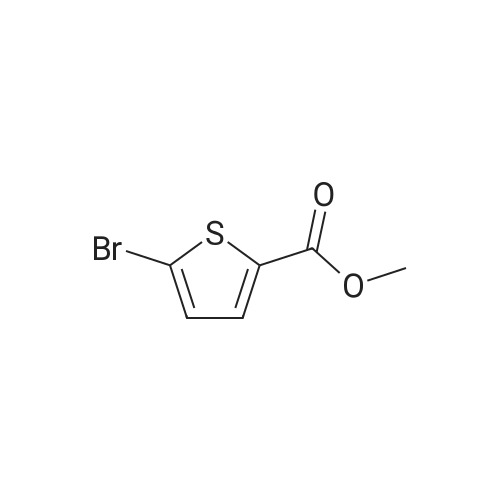
To a 1 L round bottom flask was added aluminum chloride (1 1.32 g, 85 mmol) and methyl delta-chloro^-thiophenecarboxylate (10 g, 56.6 mmol) dissolved in CHCI3 (250 ml_). Br2 (4.08 ml, 79 mmol) was added dropwise over 10 minutes. After stirring for 6 h at 25 0C, the light orange reaction solution was washed with sat NaHCO3. The organic layer was dried Na2SO4, filtered and concentrated. The residue was purified on silica gel [hexanes/EtOAc, 9:1 ] to give the product [12 g, 80%] as a white solid. LCMS (ES) m/z 256 (M+H)+ .
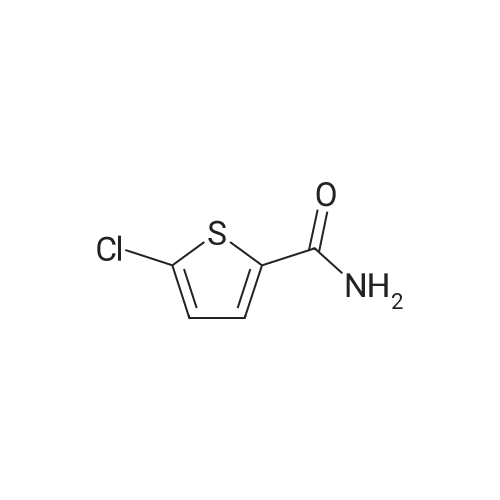
With ammonium hydroxide; In n-heptane; at 20℃; for 36h;Industry scale;
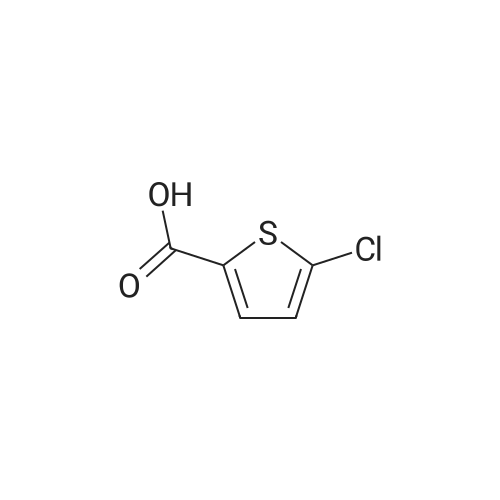
Example 3 Preparation of Compound III-B The 200-gallon reactor was charged with methanol (135 kg, 14.3 volumes), 5-chlorothiophene-2-carboxylic acid III-G (12 kg, 73.81 mol, 1.0 equiv), and sulfuric acid (6.7 kg, 68.31 mol) under nitrogen. The contents were warmed to reflux (64 C.) for 16 hours and reaction completion monitored by HPLC. The reactor was cooled to 40 C. and the mixture was vacuum distilled to an oil at <50 C. The methyl ester obtained was cooled to 20 C. and ammonium hydroxide (157 kg, 2586 mol, 35.2 equiv) was charged along with heptane (10 kg) to the reactor. The reactor contents were mixed for 36 hours at ambient temperature. The completion of the reaction was monitored by HPLC for the disappearance of methyl ester intermediate. The precipitated solids were centrifuged and washed with water followed by heptane. The isolated solids were dried at 45 C. for 14 hours to afford Compound III-B (8.42 kg, 77.1% yield).
With ammonium hydroxide; In n-heptane; at 20℃;Industry scale;
Example 3; Preparation of Compound G The 200-gallon reactor was charged with methanol (135 kg), 5-chlorothiophene-2-carboxylic acid F (12 kg, 73.81 mol, 1.0 equiv), and sulfuric acid (6.7 kg, 68.31 mol) under nitrogen. The contents were warmed to reflux (64 C.) for 16 hours and reaction completion monitored by HPLC. The reactor was cooled to 40 C. and the mixture was vacuum distilled to an oil at <50 C. The methyl ester obtained was cooled to 20 C. and ammonium hydroxide (157 kg, 2586 mol, 35.2 equiv) was charged along with heptane (10 kg) to the reactor. The reactor contents were mixed for 36 hours at ambient temperature. The completion of the reaction was monitored by HPLC for the disappearance of methyl ester intermediate. The precipitated solids were centrifuged and washed with water followed by heptane. The isolated solids were dried at 45 C. for 14 hours to afford Compound G (8.42 kg, 77.1% yield).
With ammonium hydroxide; In n-heptane; at 20℃; for 36h;Industry scale;
Example 3: Preparation of Compound GH2S04 NH3 (28% aq) Q C ^,S C02H MeOH ClySyC02Me Heptane/r.t, 37hrs S JL64C for l 6 rJs 71.1 % yieldF G[0134] The 200-gallon reactor was charged with methanol (135 kg), 5-chlorothiophene- 2-carboxylic acid F (12 kg, 73.81 mol, 1.0 equiv), and sulfuric acid (6.7 kg, 68.31 mol) under nitrogen. The contents were warmed to reflux (64 C) for 16 hours and reaction completion monitored by HPLC. The reactor was cooled to 40 C and the mixture was vacuum distilled to an oil at < 50 C. The methyl ester obtained was cooled to 20 C and ammonium hydroxide (157 kg, 2586 mol, 35.2 equiv) was charged along with heptane (10 kg) to the reactor. The reactor contents were mixed for 36 hours at ambient temperature. The completion of the reaction was monitored by HPLC for thedisappearance of methyl ester intermediate. The precipitated solids were centrifuged and washed with water followed by heptane. The isolated solids were dried at 45C for 14 hours to afford Compound G (8.42 kg, 77.1% yield).






 Chemistry
Chemistry
 Pharmaceutical Intermediates
Pharmaceutical Intermediates
 Inhibitors/Agonists
Inhibitors/Agonists
 Material Science
Material Science







 For Research Only
For Research Only
 120K+ Compounds
120K+ Compounds
 Competitive Price
Competitive Price
 1-2 Day Shipping
1-2 Day Shipping