| 87% |
With lithium aluminium tetrahydride; In tetrahydrofuran; at -78℃; for 1h; |
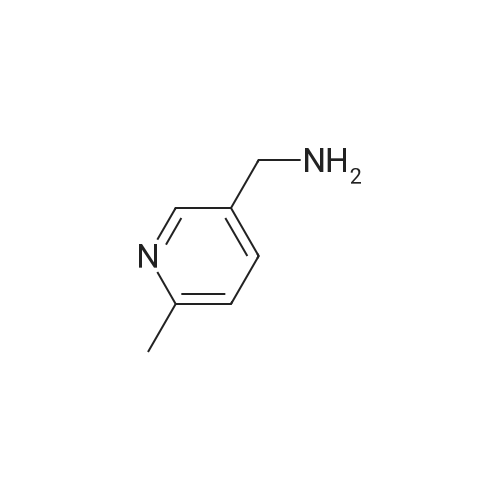
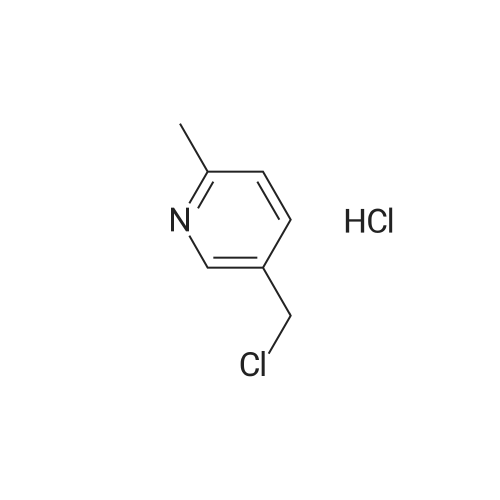
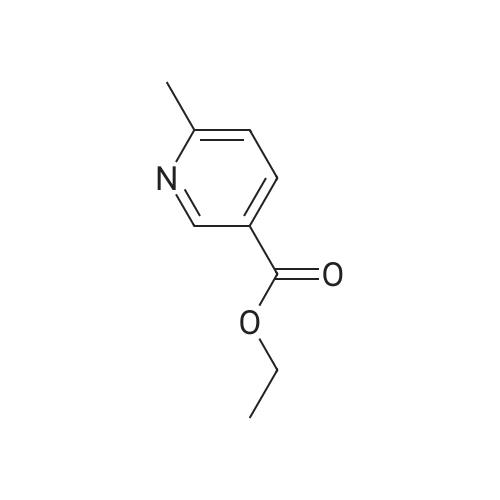
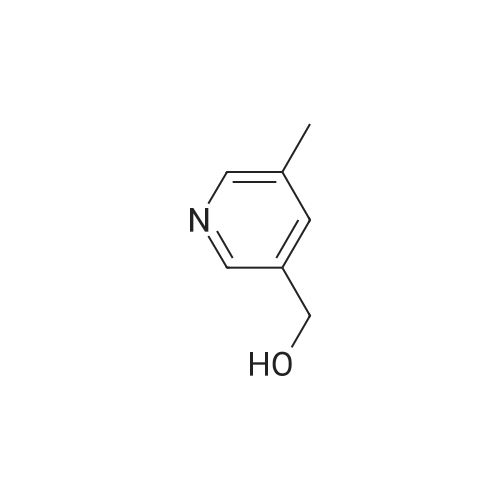
To a solution of 90 (5.0 g, 3.31 mmol) in THE was cooled to -78C, followed by addition of LAH solution (2M in THE, 4.13 mL, 8.27 mmol) slowly. The resulting mixture was stirred at -78C for 1 h. After completion of reaction (TLC monitoring), water (4.0mL) and 15% NaOH solution (4 mL) were added slowly. The resulting reaction mixture was filtered through Celite and washed with EtOAc (2 times). The organic layer was dried over anhydrous Na2SO4, filtered, and concentrated under reduce pressure to yield 91(3.5 g, 87%) as a light yellowish liquid. 1H-NMR (400 MHz, DMSO-d6): oe 8.36 (s,1H), 7.58 (d, J=8.0 Hz, 1H), 7.18 (d, J=8.0 Hz, 1H), 5.21 (t, J= 5.6 Hz, 1H), 4.46 (d,J=5.6 Hz, 2H) and 2.45 (s, 3H). LC-MS: 124.06 (M-H). To an ice-cold solution of 43(1.0 eq) and 91 (1.5 eq) in DCM was added DIAD (3.0 eq) and TPP (3.0 eq). Themixture was stirred at RT for 16h. After completion of reaction, the mixture was dilutedwith water and extracted with DCM (3 times). The combined organics was washed with brine, dried over anhydrous Na2SO4, filtered and concentrated under reduced pressure. The product was purified over silica gel column chromatography, eluting with 10% EtOAc in hexanes to yield 92 (1.3 g, 31%) as a light yellow solid. 1H-NMR (400 MHz,CDCI3): oe 8.59 (5, 1H), 7.64-7.68 (m, 2H), 7.46-7.47 (m, 1H), 7.14-7.18 (m, 2H), 5.10 (5, 2H), 3.85 (5, 3H), 2.58 (5, 3H) and 2.34 (5, 3H). MS: 272.16 (M+H). This material was then converted to 93 using the same conditions as described for 89. Analtyical data for 93: 1H-NMR (400 MHz, DMSO-d6): oe 12.90 (br s, 1H), 8.88 (5, 1H), 8.55 (5, 1H), 7.76-7.77 (m, 1H), 7.54 (5, 1H), 7.46 (d, J=7.6 Hz, 1H), 7.28 (d, J=7.6 Hz, 1H),5.17 (5, 2H), 2.47 (5, 3H) and 2.22 (5, 3H). LC-MS: 258.14 (M+H). |
| 85% |
|
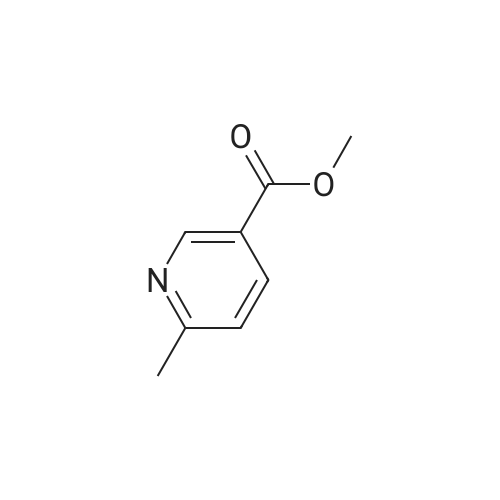
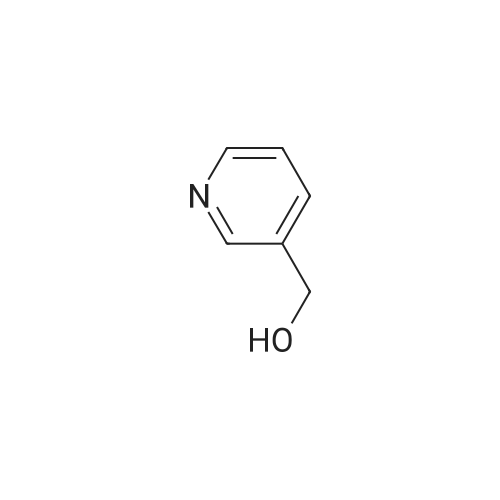
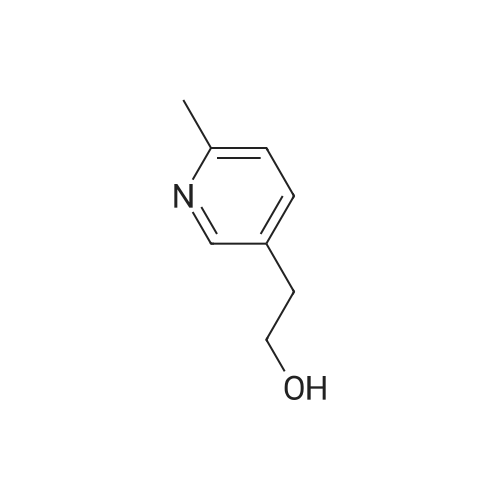
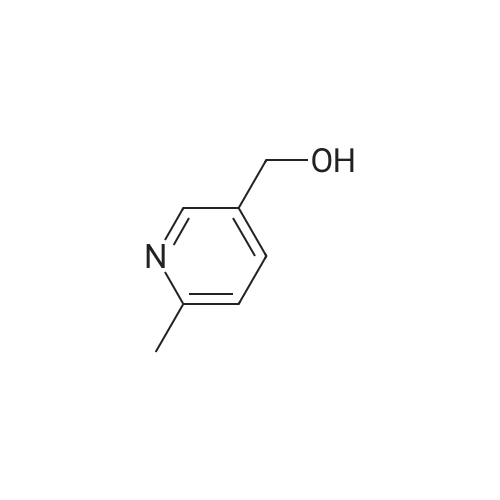
Intermediate G:; Methyl (2E)-3-r6-(Hvdroxyrnethyl)pyridin-3-yllacrylic acid <n="28"/>The title compound was prepared by the following methodology:LiAIH4, Et2O MnO2, DCM Stage 1 Stage 2 MeO OMe Stage 3K2CO3, H2OTFAA, THF mCPBA, DCM Stage 5 Stage 4 Intermediate G; Stage 1 - Preparation of (6-methylpyridin-3-yl)methanol; To a suspension of lithium aluminium hydride (7.24g, 191mmol) in Et2O (400ml) at -780C was added via cannula over a period of one hour a solution of methyl 6-methylnicotinate (19.62g, 130mmol) in Et2O (20OmL). Once addition was completed, the mixture was stirred for a further 3h. Excess lithium aluminium hydride was quenched by dropwise addition of EtOAc (40 ml). The mixture was then warmed using a water-ice bath, and further quenched with sat NH4CI (50OmL). The ethereal layer was decanted, and EtOAc was added (50OmL). The mixture was stirred vigorously, and the organic layer was again decanted. The extraction procedure was repeated twice (50OmL EtOAc). The combined organic extracts were dried (MgSO4) and concentrated to yield the desired product (13.6g, 85%). 1H NMR (300MHz, CDCI3) delta: 8.39 (1 H, s), 7.62 (1 H, dd, J=2.1 , 8.1 Hz), 7.14 (1 H, d, J=7.8Hz), 4.68 (2H, s), 2.54 (3H, s). |
| 83% |
|
a) 6-methyl-3-pyridylcarbinol Following the procedure of Example 2(b), except substituting methyl 6-methylnicotinate for N-cyclopropylmethyl isobutyramide, the title compound was prepared as a yellow oil (4.32 g, 83%). MS (ESI): 123.8 (M+H)+. |
| 82% |
|
A solution of methyl 6-methylnicotinate (50 g, 0.33 mol) in anhydrous tetrahydrofuran (100 ml) was slowly added dropwise to a suspension of LAH (12.6 g, 0.33 mol) in anhydrous tetrahydrofuran (500 ml) over a period of 30 minutes while cooling on ice, and the mixture was stirred at the same temperature for 1 hour and 20 minutes. After confirming completion of the reaction by thin layer chromatography, H2O (25 ml) was slowly added dropwise over a period of 30 minutes while cooling on ice, and the stirring was continued at room temperature for 30 minutes. Magnesium sulfate was added for drying, and the precipitate was filtered through celite and washed three times with ethyl acetate. The solvent was distilled off under reduced pressure to yield the title compound (33.6 g, 82%) as a yellow oil.1H-NMR(CDCl3)delta(ppm) 2.55(3H.s), 4.69(2H.brs), 7.16(1H.d.J=8.4Hz), 7.61(1H.dd.J=8.4and2.4Hz), 8.46(1H.d.J=2.4Hz). |
| 64% |
|
Step A: Preparation of (6-methylpyridin-3-yl)methanol: A solution of methyl6-methylnicotinate (16.3 g, 108 mmol) in MeOH (150 mL) was treated with sodium borohydride (12.2 g, 323 mmol) at ambient temperature in portions. The mixture was quenched with water (100 mL) and concentrated. This mixture was diluted with water (300 mL) and extracted with EtOAc. The combined organic extracts were dried (phase separator silicone treated filter paper) and concentrated to give (6-methylpyridin-3-yl)methanol (8.5 g, 64% yield) as a light yellow oil. |
| 62% |
|
Preparation of Reagent 3-(6-(chloromethyl)pyridin-3-yl)isoxazole; A. Preparation of (6-methylpyridin-3-yl)methanol; To a solution of lithium aluminum hydride (1 M in diethyl ether, 80 mL, 80 mmol) in 20 mL THF at -78 C. under argon, a solution of methyl 6-methylnicotinate (6.05 g, 40 mmol) in 60 mL diethyl ether was added over 1 h. The resulting reaction mixture was stirred at -78 C. for 1 h before 12 mL EtOAc was added over 10 min. The reaction mixture was allowed to warm up to 0 C. and 12 mL water was added drop-wise over 10 min. The resulting mixture was stirred for 30 min, then filtered through Celite. The filtrate was dried (Na2SO4), filtered and concentrated to obtain 3.07 g (62%) of the title compound as an off-white solid. HPLC: retention time=0.19 min. |
| 62% |
|
A. Preparation of (6-methylpyridin-3-yl)methanol To a solution of lithium aluminum hydride (1 M in diethyl ether, 80 mL, 80 mmol) in 20 mL THF at -78 C. under argon, a solution of methyl 6-methylnicotinate (6.05 g, 40 mmol) in 60 mL diethyl ether was added over 1 h. The resulting reaction mixture was stirred at -78 C. for 1 h before 12 mL EtOAc was added over 10 min. The reaction mixture was allowed to warm up to 0 C. and 12 mL water was added drop-wise over 10 min. The resulting mixture was stirred for 30 min, then filtered through Celite. The filtrate was dried (Na2SO4), filtered and concentrated to obtain 3.07 g (62%) of the title compound as an off-white solid. HPLC: retention time=0.19 min. |
|
In tetrahydrofuran; water; |
Reference Example 5 A solution of methyl 6-methylnicotinate (45.4 g) in tetrahydrofuran (50 ml) was added dropwise to a suspension of lithium aluminium hydride (LiAlH4, 5.7 g) in tetrahydrofuran (250 ml) at room temperature. The mixture was stirred at room temperature for additional 1 hour and water (30 ml) was added dropwise with ice-cooling. Insoluble materials were filtered off and the filtrate was concentrated. The residue was distilled under reduced pressure to obtain 6-methyl-3-pyridylmethanol (31 g, yield: 84%), b.p. 98-100 C./0.5 mmHg. |
|
In tetrahydrofuran; water; |
Step B: Preparation of 2-methyl-5-hydroxymethylpyridine Methyl 6-methylnicotinate (0.05 mole) is stirred in dry tetrahydrofuran (50 ml.) at 10 C. while solid lithium aluminum hydride (0.0125 mole) is added over one hour. Water (10 ml.) is added with stirring, and the mixture is concentrated to dryness. The residue is extracted several times with hot isopropanol and the combined extracts are filtered and concentrated to dryness to give 2-methyl-5-hydroxymethylpyridine. |
|
With sodium sulfate; In tetrahydrofuran; |
Reference example 2. 6-methylpyridine-3-carboaldehyde 1.00g (6.62mmol) of methyl 6-methyl nicotinate (Lancaster) was dissolved in 20ml of tetrahydrofuran anhydrous, and then 0.25g (6.59mmol) of lithium aluminum hydride was added thereto under ice bath cooling, followed by stirring for 30 minutes. Sodium sulfate 10 hydrate was added to the reaction mixture until no foam appeared, and after stirring for 2 hours, filtration with Celite was performed. The filtrate was subjected to vacuum concentration, and the residue was purified with silica gel column chromatography (ethyl acetate) to obtain 0.80g of 6-methylpyridine-3-methyl alcohol. |
|
|
In a 500 ml round-bottomed flask at room temperature under nitrogen, methyl 6-methyl-3- pyridinecarboxylate (10 g, 66.2 mmol) was dissolved in dry tetrahydrofuran (100 ml) to give a orange solution. The mixture was then cooled down to 00C and lithium aluminum hydride (36.4 ml, 72.8 mmol) was added dropwise, keeping internal temperature below 00C. At the end of addition the ice-bath was removed and the resulting solution was stirred at room temperature for 3 hours. The mixture was slowly additioned with 2.73 ml of water, 2.73 ml of NaOH 1 M and 8.2 ml of water. The resulting yellow suspension was stirred at room temperature for -30 minutes and then filtered over a Gooch funnel. The solid was washed with Et2O (3 x 100 ml). The combined organics were dried over Na2SO4, filtered and concentrated to give the title product (7.48 g) as orange oil. |
|
With lithium aluminium tetrahydride; In tetrahydrofuran; at 0℃; for 1.5h; |
To a slurry of lithium aluminum hydride (500 mg, 13.2 mmol) in anhydrous tetrahydrofuran (45 mL) at 0 C. under nitrogen was added dropwise a solution of methyl 4-methylnicotinate (15, 1.0 g, 6.6 mmol) in anhydrous tetrahydrofuran (5 mL). After 1.5 h the suspension was diluted with water (1 mL) and 6 N NaOH solution (5 mL). The precipitate was removed by vacuum filtration and the filtrate was dried over Na2SO4, filtered, and the solvent was removed under reduced pressure to provide the title compound as a light yellow oil: 1H NMR (300 MHz) 8.34 (s, 1H), 7.60 (d, J=7.9 Hz, 1H), 7.12 (d, J=7.9 Hz, 1H), 4.65 (s, 2H), 4.18-4.10 (bs, 1H), 2.51 (s, 3H) ppm. |
|
|
To aluminum lithium hydride (2.0 g) in THF (156 ml) was added dropwise methyl 6-methylnicotinate (7.8 g) in THF (78 ml) at 0C. The mixture was stirred for 2 hours at room temperature at 0C, and water (7.8 ml), 15% aqueous solution of sodium hydroxide (7.8 ml), and water (23.4 ml) were sequentially added to the mixture. The mixture was filtered with Celite, and washed with methanol. The solvent was removed under reduced pressure, and the obtained residue was purified by silica gel column chromatography, to give (6-methyl-3-pyridinyl)methanol (6.3 g)· 1H-NMR (200 MHz, CDCl3) delta 2.50 (3H, s), 4.64 (2H, s), 7.13 (1H, d, J = 8.0 Hz), 7.58 to 7.64 (1H, m), 8.34 (1H, s) |
|
With lithium aluminium tetrahydride; In tetrahydrofuran; at 0 - 20℃; for 2h; |
Reference Example 18 Production of (6-methylpyridin-3-yl)methanol To a solution of methyl 6-methylnicotinate (2.45 g) in tetrahydrofuran (30 ml) was added slowly lithium aluminum hydride (0.62 g) at 0 C. After the temperature of the reaction mixture was elevated to room temperature, the reaction mixture was stirred at the same temperature for 2 hours.. The reaction mixture was poured into saturated brine, and extracted with ethyl acetate.. The ethyl acetate layer was dried over magnesium sulfate and concentrated.. The residue was purified by column chromatography (carrier: silicagel, eluant: hexane-ethyl acetate) to obtain the titled compound (1.49 g) as a yellow oily substance.1H-NMR (CDCl3) delta 2.51 (3H, s), 3.86 (1H, brs), 4.66 (2H, s), 7.13 (1H, d, J=8.0 Hz), 7.61 (1H, dd, J=2.2, 8.0 Hz), 7.63 (1H, d, J=2.2 Hz). |
|
|
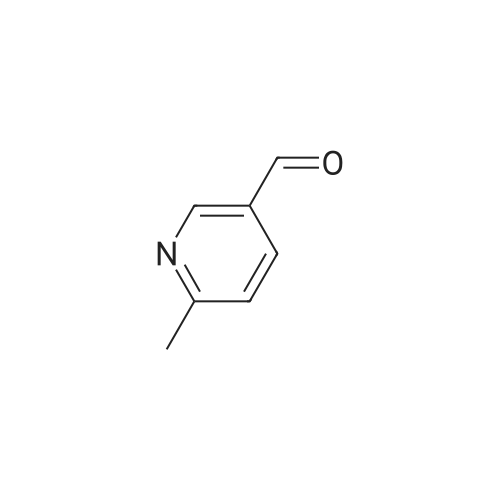
A solution of methyl 6-methylnicotinate (0.5 g, 3.3 mmol) in THF (16 mL) at 0 C. was treated dropwise with lithium aluminum hydride in THF (6.6 mL, 1 M), stirred at 0 C. for 1.5 hours, treated with ethyl acetate (3 mL), stirred at 25 C. The reaction was partitioned between ethyl acetate and saturated NaHCO3, and the organic phase was washed with brine and dried over MgSO4, filtered and concentrated. A solution of the residue (0.395 g) in dichloromethane (16 mL) was treated with MnO2 (2 g), stirred at 25 C. for 68 hours, filtered through celite to give the title compound (0.326 g, 80% yield), which was used without further purification. |
|
|
Intermediate 1 Ethyl (2£)-3-(5-formylpyridin-2-yl)acrylateStage 1; Lithium aluminium hydride (23g, 1.2eq) in THF (50OmL) was cooled to -78C. Methyl-6- methylnicotinate was dissolved in THF (20OmL) and charged to the reaction at below - 700C. The reaction was allowed to warm to 00C over 1h and aged at ~0C for 1 h. On completion the reaction was quenched with sat. NaHCO3 (25OmL) below 10C.The reaction mixture was filtered to remove inorganics, the filter cake was washed with THF and the filtrate concentrated in vacuo to remove most of the THF. The residue was separated between ethyl acetate and water, the aqueous layer being extracted three times with EtOAc. Combined organics washed with K2CO3(aq), dried (MgSO4) then concentrated to dryness to afford the product (40.5g). 1H NMR (CDCI3): (8.35 ,1 H,s), 7.61 (1 H,dd), 7.13 (1 H,d), 4.65 (2H,d), 2.51 (3H,d). A second extraction of the aqueous layer provided additional product (5.5g) which was combined with the product from the first extraction. |
|
With hydrogenchloride; diisobutylaluminium hydride; In methanol; ethyl acetate; toluene; |
Preparation of 5-hydroxymethyl-2-methyl-pyridine 5-Methoxycarbonyl-2-methyl-pyridine (3.0 g, 19.8 mmole) was dissolved in toluene (60 ml) and cooled to -78 C. To this solution, a 39.7 ml of a solution of 1M diisobutylaluminum hydride in toluene was added dropwise. The reaction mixture was stirred for 1 hour at -78 C., allowed to warm to room temperature and stirred for an additional 18 hours. Methanol (8 ml) was then added dropwise at room temperature to the reaction mixture. The mixture was stirred until a thick gel was formed. 1N HCl (10 ml) was added to dissolve the gel. The resulting phases were separated, the organic phase was extracted in ethyl acetate and dried over MgSO4. The solvents were removed yielding crude 5-hydroxymethyl-2-methylpyridine (540 mg) as an oil, which was used without further purification. |
|
|
A solution of methyl 6-methylnicotinate (30 g, 198 mmol) in dry THF (150 mL) was added dropwise into a suspension of LiAlH4 (11 g, 289 mmol) in THF (200 mL) at 0 to - 5 C. The grey suspension was stirred at RT for lh. The reaction mixture was cooled to -5 to 0 C and water (11 mL) was added dropwise under nitrogen followed by slow addition of 15% NaOH (11 mL) and water (33 mL). The reaction was warmed to RT and stirred for 30 min. at RT. The resulting suspension was filtered and the filtrate was concentrated under vacuum to give (6-methylpyridin-3-yl)methanol as a yellow oil (23 g). |
|
With lithium aluminium tetrahydride; In tetrahydrofuran; at -5 - 5℃; for 2h; |
(6-Methylpyridin-3-yl)methanolThe (6-Methylpyridin-3-yl)methanol used in the above process was prepared as follows:THF (4 L) was added to a flask containing LiAIH4 (150.4 g, 3.96 mol) and the resulting suspension cooled to < 0 C. Methyl 6-methylnicotinate (500 g, 3.31 mol) in THF (1 L) was added dropwise to the reaction over 2.5 h, maintaining a temperature < 5 C. Stirring was continued at 0+/- 5 C for 2 h and then the reaction was quenched slowly over 1 h 25 min with satd. NaHC03 (1 L) at < 10 C. The reaction was then stirred overnight at ambient temperature. The reaction was then filtered, and the filter cake washed with EtOAc (2.5 L) and the filtrate concentrated to dryness. The residue was partitioned between NaHC03 (500 ml_) and EtOAc (1 .5 L). The layers were separated and the aqueous layer extracted with EtOAc (3 x 1 L). The combined organic layers were concentrated to dryness. The residue was dissolved in EtOAc (1 L), dried (Na2S04) and concentrated to dryness to give the title compound (366 g). 1H NMR (CDCI3): 8.31 (1H, d, J=2.1 ), 7.59 (1 H, dd, J=7.8, 2.1 ), 7.10 (1 H, d, J=7.8), 4.69 (2H, s), 2.48 (3H, s). |
| 0.61 g |
With lithium triethylborohydride; In tetrahydrofuran; at -78 - 0℃; |
b) (6-methylpyridin-3-yl)methanol 1 M Lithium triethylborohydride (super hydride) in THF (12.0 mL, 0.01 moles) was added to a stirred solution of methyl 6-methylnicotinate (0.9 g, 0.06 moles) as obtained in step a) in dry THF (10 mL) at -78C. The reaction mixture was slowly allowed to rise to 0 C and stirred for 1 .5 h. Once the starting material was consumed (monitored by TLC), the reaction mixture was quenched with saturated aqueous ammonium chloride solution and extracted with ethyl acetate (2 x 100 mL). The organic layer was washed with water, dried over anhydrous Na2S04 and concentrated under reduced pressure. Purification by column chromatography (silica gel, 3% MeOH in dichloromethane) provided the title compound as pale yellow solid (0.61 g, 84%). 1 H NMR (400 MHz, DMSO): delta 8.37 (s, 1 H), 7.57 (dd, J = 2.0 Hz & J2 = 7.9 Hz; 1 H), 7.19 (d, J = 7.8 Hz; 1 H), 5.21 (t, J = 5.9 Hz; 1 H), 4.47 (d, J = 5.4 Hz; 2H), 2.43 (s, 3H); MS (ES-MS): m/z 124.0 (M + 1 ). |
|
With lithium aluminium tetrahydride; In tetrahydrofuran; at 0 - 20℃; for 1h; |
Step 1: Intermediate 31-b To a solution of methyl 6-methylnicotinate 31-a (20.10 g, 133 mmol) in THF (90 ml) cooled to 0C was added drop wise a 1.0 M solution of LiAIH4 in THF ( 100 ml, 100 mmol) and the reaction was then stirred at 0C for 1 hour. Water (3.8 ml) was slowly added, followed by 15% NaOH (3.5 ml) and water (11.4 ml) and the mixture was stirred at room temperature for 1 hour. The reaction was filtered over celite and volatiles were removed in vacuo to provide intermediate 31-b as a yellow oil. |
|
With lithium aluminium tetrahydride; In tetrahydrofuran; at 0℃; for 1h; |
Step 1: Intermediate 31-b [0181] To a solution of methyl 6-methylnicotinate 31-a (20.10 g, 133 mmol) in THF (90 ml) cooled to 0 C. was added drop wise a 1.0 M solution of LiAlH4 in THF (100 ml, 100 mmol) and the reaction was then stirred at 0 C. for 1 hour. Water (3.8 ml) was slowly added, followed by 15% NaOH (3.5 ml) and water (11.4 ml) and the mixture was stirred at room temperature for 1 hour. The reaction was filtered over celite and volatiles were removed in vacuo to provide intermediate 31-b as a yellow oil. |
|
|
To a solution of methyl 6-methylnicotinate (25 g, 0.16 mol) in ethyl ether (620 mL) under nitrogen was added dropwise sodium bis(2-methoxyethoxy)aluminium hydride (Red-Al) (65wt.% in toluene, 110 mL, 0.37 mol) at room temperature. The mixture was then heated to reflux for 1.5 hr. After cooling, the reaction mixture was quenched with water (500 mL) at 0 0C and the aqueous layer was extracted with EtOAc. The combined organic layers were dried over anhydrous MgSO4, filtered, and concentrated. The residue was purified by silica gel column to afford the title compound. |
|
|
To a solution of methyl 6-methylnicotinate (5.0 g, 33 mmol) in THF (120 mL) under argon at -78 0C was added diisobutylaluminium hydride (IM in hexane, 66 mL, 66 mmol). The reaction mixture was allowed to warm to r.t. and stirred for 4 days. DCM (120 mL) was added. The mixture was added to a saturated aqueous solution of Rochelle salt (150 mL) at 0 0C. The mixture was stirred until two layers had formed. The phases were separated and the aqueous phase was extracted with DCM (2 x 150 mL). The combined organic phases were dried (MgSO4) and concentrated in vacuo to give (6-methyl-pyridin-3-yl)-methanol as a yellow oil, which was used without further purification in the next step. Analytical LCMS: purity 83% (System A, Rtau = 0.44 min), ES+: 123.9 [MH]+. |






 Chemistry
Chemistry
 Pharmaceutical Intermediates
Pharmaceutical Intermediates
 Inhibitors/Agonists
Inhibitors/Agonists
 Material Science
Material Science







 For Research Only
For Research Only
 120K+ Compounds
120K+ Compounds
 Competitive Price
Competitive Price
 1-2 Day Shipping
1-2 Day Shipping