* All experimental methods are cited from the reference, please refer to the original source for details. We do not guarantee the accuracy of the content in the reference.
With potassium carbonate; In N-methylpyrrolidone (NMP); at 0 - 120℃; for 6h;
A solution of <strong>[327056-73-5]3-chloro-5-fluorobenzonitrile</strong> (59a, 9.2 g, 59.14 mmol), 6 (10.44 g, 53.76 mmol), K2CO3 (22.29 g, 161.3 mmol) and NMP (100 mL) was stirred and heated to 120 C. for 6 h. The reaction mixture was cooled to 0 C. and diluted with saturated NaHSO3 (100 mL) and twice extracted with EtOAc. The combined organic extracts were washed sequentially with water (6 times) and brine, dried (Na2SO4), filtered and concentrated in vacuo. The crude product was purified by SiO2 column chromatography eluting with a EtOAc/hexane gradient (0 to 10% EtOAc) to afford 9.28 g (48% theory) of 112a.
A solution of methyl 3-hydroxy-5-methoxy-phenyl acetate (32b, 0.500 g, 2.55 mmol), <strong>[327056-73-5]3-chloro-5-fluorobenzonitrile</strong> (0.3964 g, 2.55 mmol), K2CO3 (1.057 g, 7.65 mmol) and NMP (5 mL) were stirred and heated at 120 C. for 12 h. The reaction mixture was cooled to RT and diluted with H2O (35 mL), adjusted to pH 11 with 1N NaOH and twice extracted with EtOAc. The combined organic extracts were washed sequentially with water (6 times) and brine, dried (Na2SO4), filtered and concentrated in vacuo. The crude product was purified by SiO2 column chromatography eluting with a EtOAc/hexane gradient (0 to 25% EtOAc) to afford 34.
A solution of 28a (0.500 g, 2.77 mmol), <strong>[327056-73-5]3-chloro-5-fluoro-benzonitrile</strong> (0.4316 g, 2.77 mmol), K2CO3 (1.150 g, 8.32 mmol) and NMP (5 mL) was stirred and heated for 8 h at 120 C. The reaction mixture was cooled to RT and diluted with H2O (150 mL), acidified with 10% aqueous HCl and twice extracted with EtOAc. The combined organic extracts were washed sequentially with water (6 times) and brine, dried (Na2SO4), filtered and concentrated in vacuo. The crude product was purified by SiO2 column chromatography eluting with EtOAc/hexane gradient (0 to 25% EtOAc) to afford 30a.
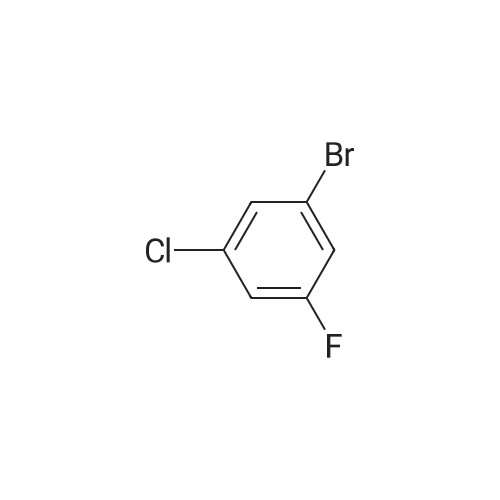
A mixture of 1-bromo-3-chloro-5-fluorobenzene (8Og, 480mmol), zinc cyanide (33.65g, 290mmol) and zinc dust (0.94g, 14.46mmol) in DMF (34OmL) was stirred at rt for 5 min. Dichloro[1 ,1 '- EPO <DP n="34"/>bis(diphenylphosphino)ferrocene] palladium(ll) (4.99g, 16mmol) was then added and the mixture was heated under reflux for 50 min. The reaction mixture was cooled to rt and filtered through Arbocel, washing through with diethyl etherpentane (50:50, 7 x 10OmL). The phases were separated and the organic phase was diluted with water (10OmL) and extracted with further diethyl ethe?pentane (50:50, 3 x 10OmL). The combined organic solutions were then washed with water, dried over magnesium sulfate and concentrated in vacuo. Distillation of the residue under reduced pressure afforded the title compound as a colourless solid in 66% yield, 48.5g. LRMS: m/z APCI 155 [MH+]
With potassium carbonate; In dimethyl sulfoxide; at 80℃; for 20h;
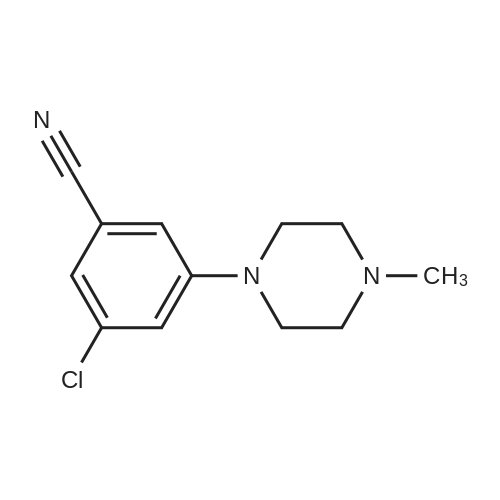
5-Fluoro-3-chloro-benzonitrile (Ig, 6.4 mmol) was dissolved in DMSO (20 ml) followed by addition Of K2CO3 (1.3g, 9.6 mmol) and 1 -methyl piperazine (1.4 ml, 12.8 mmol). The reaction mixture was heated at 80 0C for 20 hours. Diethyl ether was added to the crude material (10 ml) then acidified with IN HCl. A precipitate was filtered off from the crude reaction mixture to give 3-chloro-5-(4-methyl- rhoirhoerazin-l-yl)-benzonitrile (1.4g, 93% yield) as a white solid (LC/MS: Rt 1.83 [M + H]+ 236, acidic method).
93%
With potassium carbonate; In dimethyl sulfoxide; at 80℃; for 20h;
18A. Synthesis of 3-Cmoro-5-(4-methyl^iperazm-l-ylVbeiizomtrile; 5-Fluoro-3-chloro-benzonitrile (Ig, 6.4 mmol) was dissolved in DMSO (20 ml) followed by addition OfK2CO3 (1.3g, 9.6 mmol) and 1-methyl piperazine (1.4 ml, 12.8 mmol). The reaction <n="215"/>mixture was heated at 80 0C for 20 hours. Diethyl ether was added to the crude material (10 ml) then acidified with IN HCl. A precipitate was filtered off from the crude reaction mixture to give 3-chloro-5-(4-methyl-piperazin-l-yl)-benzonitrile (1.4g, 93% yield) as a white solid (LC/MS: Rt 1.83 [M + H]+ 236, acidic method).
61%
With potassium carbonate; In dimethyl sulfoxide; at 100℃;
STEP A A mixture of <strong>[327056-73-5]3-chloro-5-fluoro-benzonitrile</strong> (1.1 g, 7.07 mmol), 1-methyl-piperazine (1.18 ml, 10.6 mmol), and K2CO3 (2.92 g, 21.21 mmol) in DMSO (25 ml) was heated to 100C overnight and then partitioned between water and Et2O. The aqueous phase was extracted with Et2O and the collected organic phases were dried over Na2SO4 and evaporated under vacuum. The residue was dissolved in Et2O and extracted with 0.5 M HCl. The aqueous phase was basified with NH4OH and extracted with DCM. The organic phase was dried over Na2SO4 and evaporated to give 1.01 g of 3-chloro-5-(4-methyl-piperazin-1-yl)-benzonitrile. Y=61%
With potassium carbonate; In N,N-dimethyl-formamide; at 140℃; for 0.666667h;microwave irradiation;
Preparation 4: 1 -[3-ChIoro-5-(4-methyI-piperazin-1 -yl)-phenyl]-ethanone; STEP A; A mixture of <strong>[327056-73-5]3-chloro-5-fluoro-benzonitrile</strong> (1 g, 6.45 mmol), 1-methyl-piperazine (0.715 ml, 6.45 mmol), and K2CO3 (2.64 g, 19.3 mmol) in DMF (5 ml) was heated to 1400C for 40 min in a microwave apparatus. The resulting slurry was filtered and the solvent was removed in vacuo to give 1 g of 3-chloro-5-(4-methyl- piperazin-1-yl)-benzonitrile. The crude reaction mixture was used in the next step without any further purification.
With potassium carbonate; In dimethyl sulfoxide; at 20 - 80℃;
3-Chloro-5-fluorobenzonitrile (0.50 g, 3.21 mmol) in dimethyl sulfoxide (10 ml.) was stirred at room temperature under an atmosphere of argon (following a similar method to that of WO200670195). Potassium carbonate (0.67 g, 4.82 mmol) and then 1-methylpiperidine (0.71 ml_, 6.42 mmol) were added. The reaction mixture was heated at 80 0C overnight. The reaction mixture was cooled to room temperature and diethyl ether (5 ml.) was added. A white precipitate formed which was filtered off. The filtrate was acidified using 1 M hydrochloric acid to pH 5. The white precipitate was filtered. Further product precipitated out in the filtrate and was collected. This was repeated twice to give the title compound. LC/MS (ES+ve): [M+H]+ at m/z 236, 238 (Ci2H14CIN3 requires [M+H]+ at m/z 236, 238).
To a solution of the appropriate phenol (1 eq.) in DMF (0.8 to 1.85mLmmor1) was added cesium carbonate (1-2 eq.) at rt and the solution was stirred for 10 min. The compound from preparation 37 (1.3 eq.) was then added and the reaction mixture was heated at 85 C for up to 48h (reactions monitored by tic). The solvent was removed in vacuo and the residue was partitioned between EtOAc (5OmL) and brine (5OmL). The phases were separated and the aqueous layer extracted with EtOAc (1 OmL). The organic extracts were combined, dried over magnesium sulfate and the solvent was removed in vacuo to give the crude residue. Purification by column chromatography on silica gel using pentane:ethyl acetate as eluent afforded the desired product.
Preparation 23: 3-Chloro-5-[2-methoxy-5-(trifluoromethoxy)phenoxylbenzonitrile To a solution of the compound of preparation 22 (270mg, 1.30mmol) in DMF (5mL) was added cesium carbonate (551 mg, 1.69mmol) at rt. The reaction mixture was stirred for 5 min and the compound from preparation 37 (1.69mmol, 263mg) was added. The mixture was then heated at 85 0C for 3h and cooled to rt. Brine was added followed by water and the aqueous phase was extracted with EtOAc. The organic extract was dried over magnesium sulfate and the solvent was concentrated in vacuo to afford the crude residue. Purification by column chromatography on silica gel using pentane:ethyl acetate (88:12) as eluent afforded the desired product, 360mg (81%). LRMS (APCI) 343 [MH"]; Preparations 2-7 To a solution of the appropriate phenol (1 eq.) in DMF (0.8 to 1.85mLmmor1) was added cesium carbonate (1-2 eq.) at rt and the solution was stirred for 10 min. The compound from preparation 37 (1.3 eq.) was then added and the reaction mixture was heated at 85 C for up to 48h (reactions monitored by tic). The solvent was removed in vacuo and the residue was partitioned between EtOAc (5OmL) and brine (5OmL). The phases were separated and the aqueous layer extracted with EtOAc (1 OmL). The organic extracts were combined, dried over magnesium sulfate and the solvent was removed in vacuo to give the crude residue. Purification by column chromatography on silica gel using pentane:ethyl acetate as eluent afforded the desired product. EPO <DP n="25"/>A = 4-hydroxy-3-methoxybenzonitrile prepared as described in Synthesis 1989(6); 451-2. The product was isolated after trituration with methanol.B = potassium carbonate was used in place of cesium carbonate.
The reaction was repeated and the two batches of pot material are distilled through a 430*20 mm column filled with glass helices at 70-80 and 8 mm to give 3-chloro-5-fluorobenzonitrile as a volatile white solid.
10
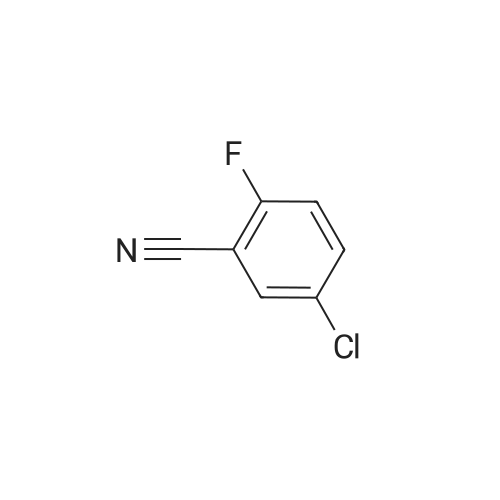
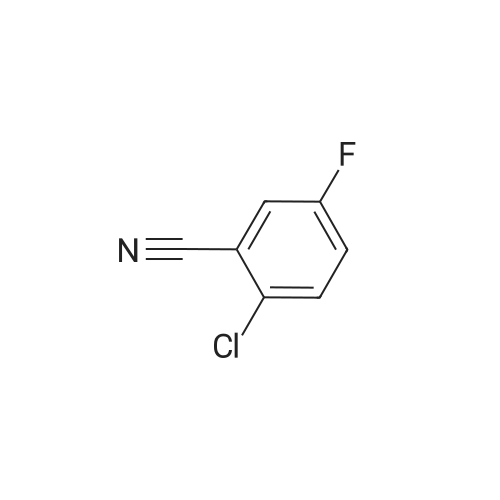
[ 327056-73-5 ]
3-chloro-5-fluorobenzamide[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
With sulfuric acid; In water;
EXAMPLE 43 A mixture of sulfuric acid (500 ml) and water (100 ml) is heated to 95 and <strong>[327056-73-5]3-chloro-5-fluorobenzonitrile</strong> (94.8 gms) added. After 4 hours the mixture is cooled and poured over ice and water added to make 3.6 L. In 1.2 L portions the mixture is filtered, the solid rinsed with hexane and the aqueous rinsed with hexane (4*120 ml) then extracted with ethyl acetate (2*100 ml). The solid and the ethyl acetate extracts were combined, dried over magnesium sulfate and stripped to give 3-chloro-5-fluorobenzamide as a white solid.






 Chemistry
Chemistry
 Pharmaceutical Intermediates
Pharmaceutical Intermediates
 Inhibitors/Agonists
Inhibitors/Agonists
 Material Science
Material Science







 For Research Only
For Research Only
 120K+ Compounds
120K+ Compounds
 Competitive Price
Competitive Price
 1-2 Day Shipping
1-2 Day Shipping