| 34%; 50% |
|
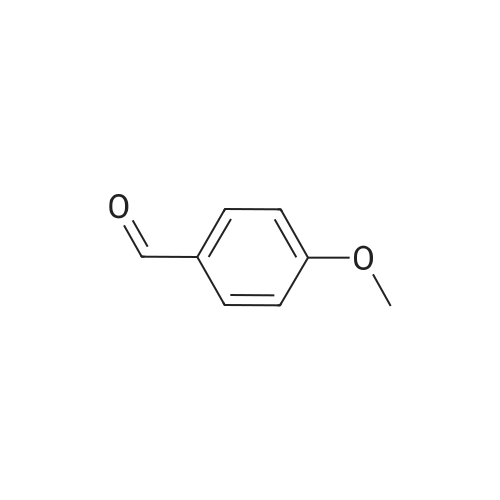
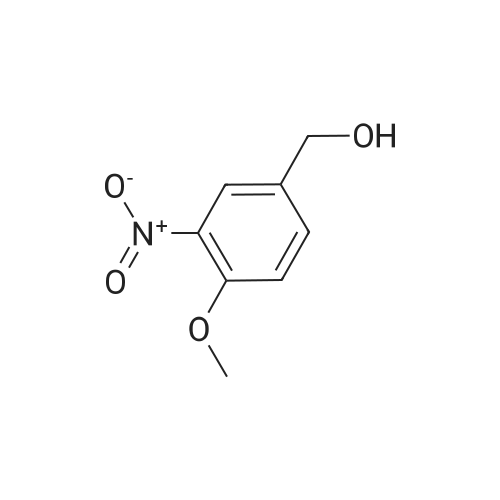
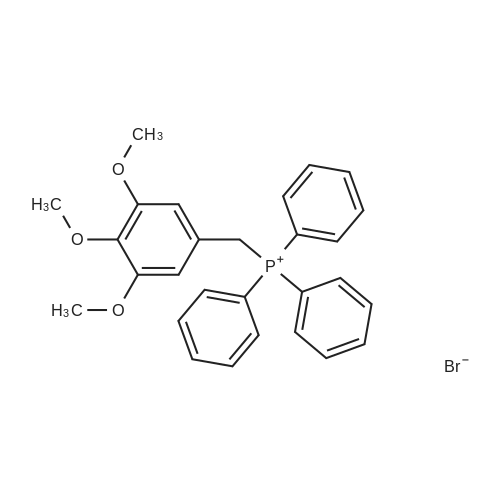
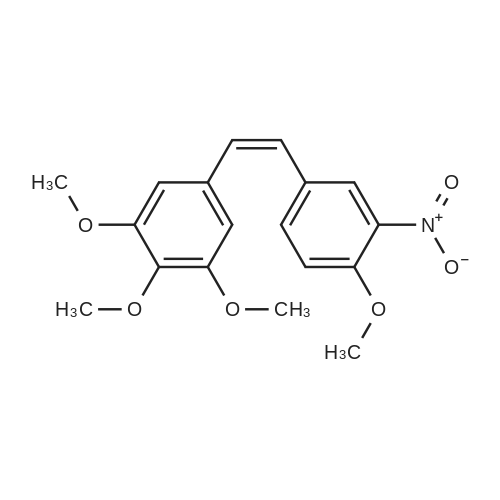
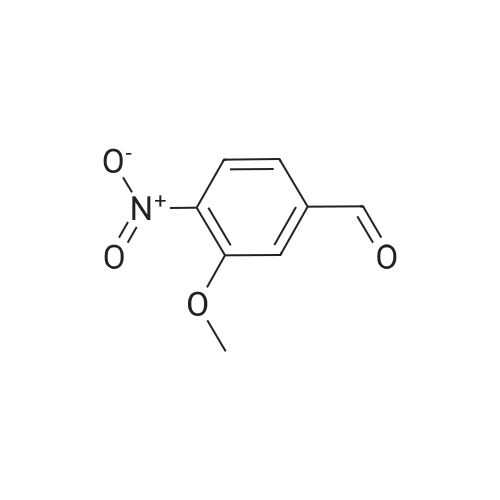
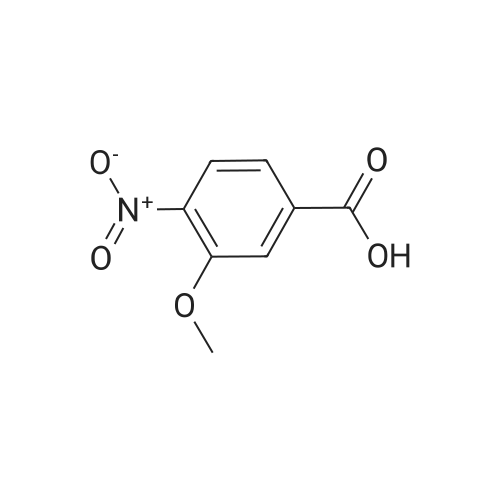
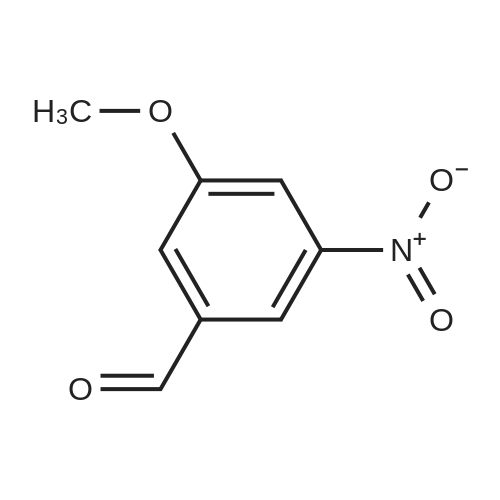
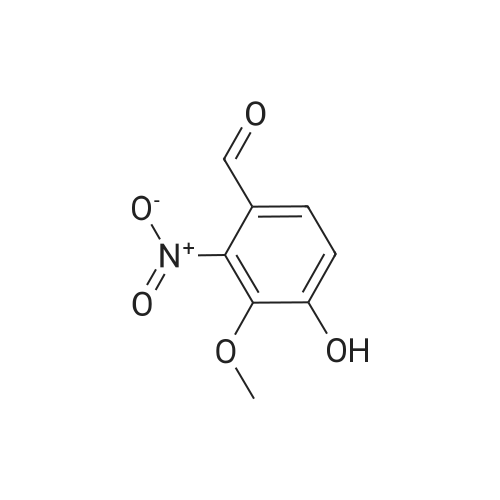
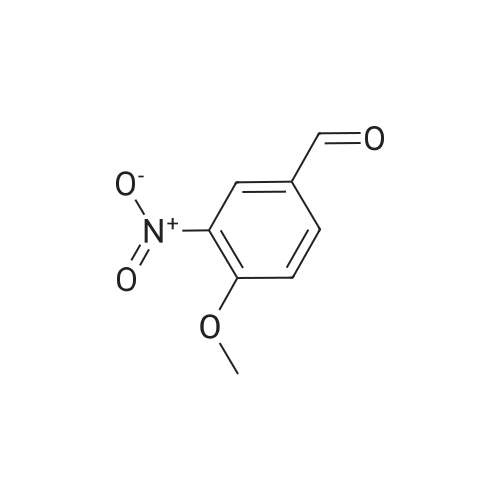
A mixture of 3,4,5-trimethoxybenzyltriphenylphosphoniumbromide [22] (1.57 g, 3 mmol), powdered potassiumcarbonate (1.38 g, 10 mmol) and 18-crown-6 (0.01 g,0.04 mmol) in dichloromethane (20 mL) was stirred at roomtemperature for 10 min. Then, <strong>[31680-08-7]4-methoxy-3-nitrobenzaldehyde</strong>(0.54 g, 3 mmol) was added and the reaction mixturewas refluxed for 6 h (monitored by TLC). The inorganicsalts were filtered after cooling and the filtrate was concentratedunder reduced pressure to give a mixture of isomers1, 2 and triphenylphosphine oxide. Recrystallization ofthe crude reaction mixture from ethanol afforded pure Estilbene1 (0.40 g, 38 % yield). Column chromatography ofthe mother liquors (petroleum ether/acetone, 15:1 v/v) gaveZ-isomer 2 (0.35 g, 34 % yield) and 1 (0.12 g, 12 % yield). (E)-5-(4-Methoxy-3-nitrostyryl)-1,2,3-trimethoxybenzene(1)Orange crystals. Rf = 0.52. Mp: 151-153 C. 1H NMR(CDCl3, 300 MHz): δ 3.87 (s, 3H, OCH3), 3.91 (s, 6H,OCH3), 3.98 (s, 3H, OCH3), 6.72 (s, 2H, ArH), 6.91 (d, 1H,J = 16.3 Hz, = CH), 6.99 (d, 1H, J = 16.3 Hz, = CH),7.08 (d, 1H, J = 8.7 Hz, ArH), 7.64 (dd, 1H, J = 1.6 Hz,J = 8.7 Hz, ArH), 7.99 (d, 1H, J = 1.6 Hz, ArH). 13CNMR (CDCl3, 75 MHz): δ 55.9; 56.5; 60.8; 103.5; 113.6;122.9; 124.9; 129.3; 130.1; 131.6; 132.1; 138.1; 139.6;151.9; 153.3.(Z)-5-(4-Methoxy-3-nitrostyryl)-1,2,3-trimethoxybenzene(2)Yellow crystals. Rf = 0.58. Mp: 122-123 C. Lit. Mp:123-124 C [12]. 1H NMR (CDCl3, 300 MHz): δ 3.70 (s,6H, OCH3), 3.84 (s, 3H, OCH3), 3.92 (s, 3H, OCH3),6.41-6.46 (m, 3H, ArH, = CH), 6.57 (d, 1H,J = 12.1 Hz, = CH), 6.94 (d, 1H, J = 8.7 Hz, ArH), 7.42(dd, 1H, J = 2.2 Hz, J = 8.7 Hz, ArH), 7.78 (d, 1H,J = 2.2 Hz, ArH). 13C NMR (CDCl3, 75 MHz): δ 55.9;56.5; 60.9; 105.8; 113.1; 125.9; 126.8; 129.7; 131.3; 131.7;134.6; 137.7; 139.4; 151.6; 153.8. |
|
With sodium hydride; In dichloromethane; at 0 - 20℃; |
General procedure: To a stirred solution of nitrobenzaldehydes (10, 11, 19, 1 mmol) and 3,4,5-trimethoxybenzyl triphenylphosphonium bromide (9, 1.1 mmol) in anhydrous dichloromethane, sodium hydride (4 mmol) was added at 0 C. The reaction mixture was stirred at room temperature for 18 h and monitored by TLC. After completion of the reaction appropriate amount of water, until foaming stopped, was added at 0 C. The organic layer was separated and the aqueous layer was extracted with chloroform. The combined organic layers were washed with brine solution, dried over anhydrous Na2SO4, filtered and the solvent was removed under reduced pressure to get crude compounds, nitro Z-and E-stilbenes isomeric mixture. The isomers were separated by using column chromatographyto give pure Z-stilbenes. |






 Chemistry
Chemistry
 Pharmaceutical Intermediates
Pharmaceutical Intermediates
 Inhibitors/Agonists
Inhibitors/Agonists
 Material Science
Material Science







 For Research Only
For Research Only
 120K+ Compounds
120K+ Compounds
 Competitive Price
Competitive Price
 1-2 Day Shipping
1-2 Day Shipping