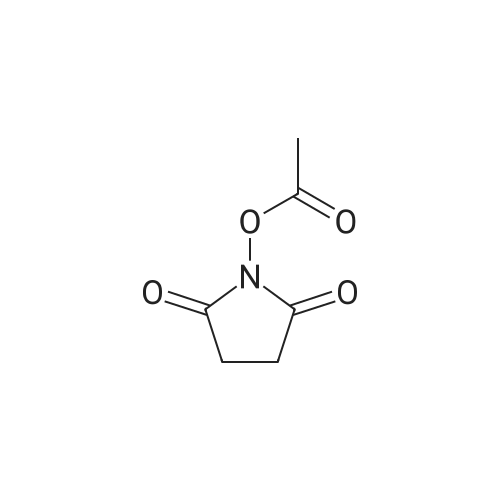
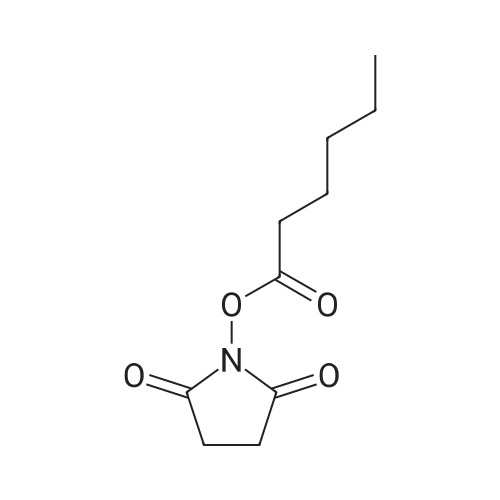
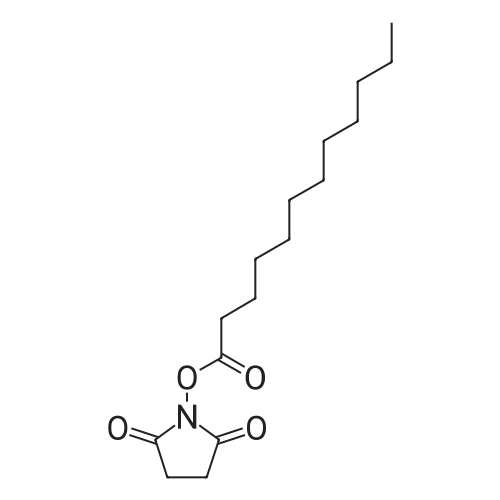
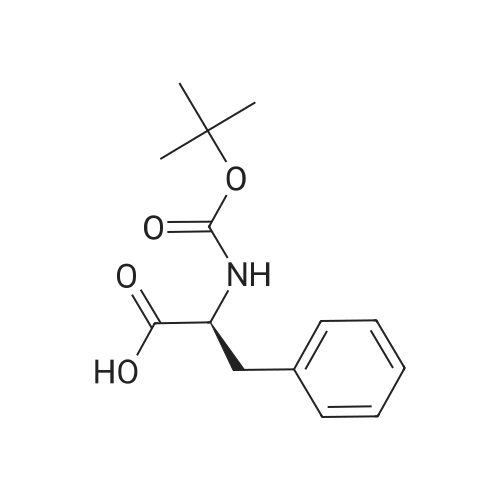
| 91% |
With triphenylphosphine; In N,N-dimethyl-formamide; at 20℃; for 10h; |
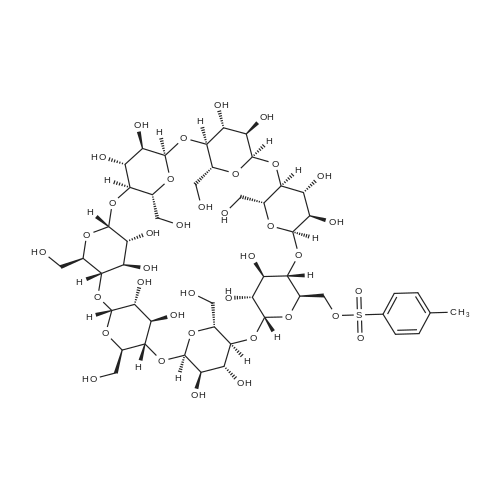
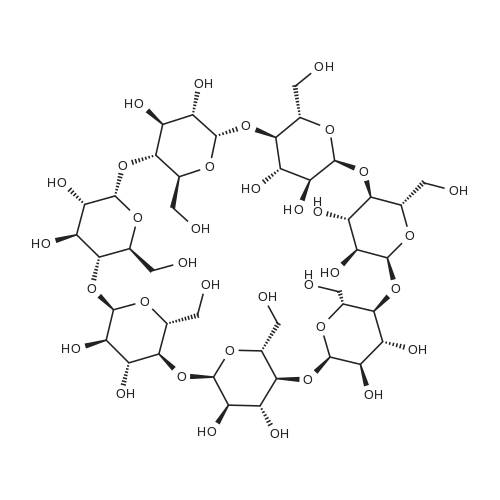
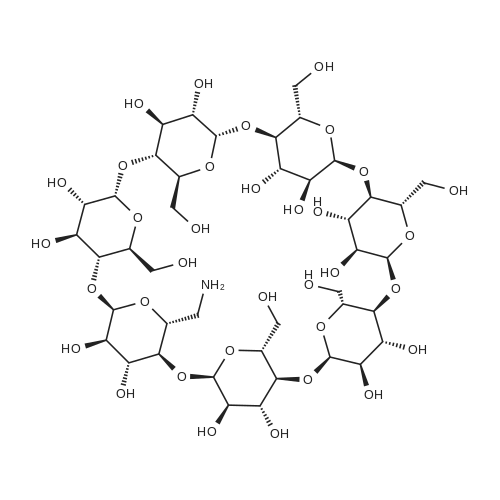
The detailed preparation for the picolinamide-modified beta-cyclodextrin/Pd(II) complex (Pd(II)PCA-beta-CD) is illustrated in Scheme 1. First, mono-6-tosyl beta-cyclodextrin (1, Tos-beta-CD) was obtained by the method of Khan etc (Khan & Pitchumani, 2016). Then, Tos-beta-CD was reacted with NaN3 to form 6-monodeoxy-6-monoazido-beta-CD (2, N3-beta-CD), which was further reduced by triphenylphosphine to give a key intermediate: 6-monodeoxy-6-amino-beta-CD (3). PCA-beta-CD (4) was prepared by condensation of PCA (2-pyridinecarboxylic acid) with NH2-beta-CD using N,N?-dicyclohexylcarbo-diimide (DCC) and 1-hydroxy-1H-benzotriazole (HOBt) in N,N?-dimethylformamide (DMF). The synthesis of PCA-(CH2)nNH-beta-CD (6) was achieved in two simple steps: nucleophilic substitution (from 1 to 5) and amidation (from 5 to 6). Finally, all the obtained ligands were stirred with Pd(OAc)2 at room temperature for 24 h in toluene, and the target complexes Pd(II)PCA-beta-CDs (C1-C3) were obtained as light yellow powders. The water solubility of the 4, 6 ligands and C1, C2, C3 complexes in different temperatures were studied. The results showed the complexes have high solubility in the aqueous solution (see Table 1S in SI). Reaction conditions: (a) TsCl, NaOH, CH3CN, H2O, rt, 2h, 15% yield; (b) NaN3, DMF, 80C, 12h, 93% yield; (c) PPh3, DMF, rt, 10h, 91% yield; (d) DCC, HOBt, DMF, 4h at 0C, 18h at rt, 72-83% yield; (e) Pd(OAc)2, toluene, rt, 24h, 60-70% yield; (f) diamine, 80C, 12h, 30% yield. |
| 90% |
With 5%-palladium/activated carbon; hydrazine hydrate; In methanol; water; for 0.333333h;Reflux; |
6-monoamino-6-monodeoxy-betaCD (Free Base) (1)11.60 g (0.01 mol) of 6-monoazido-6-monodeoxy-betaCD was dispersed cold, with stirring, in H2O:MeOH-4:1 mixture (100 mL). First a suspension of Pd/C (0.58 g, 5% Pd content in 3 mL H2O), and then hydrazine monohydrate (5 g, 5 mL, 0.1 mol) were added and heated under reflux. After 20 min with stirring under reflux, the mixture was cooled to about 50 C., filtered to remove the catalyst and washed three times with water (15 mL). After evaporation of the solvents, the crude product was precipitated in an aqueous solution (50 mL) of NH4OH 25% (2 mL). The crystals were filtered, washed three times with MeOH (10 mL) and dried at 60 C., under reduced pressure (10 mbar) overnight in the presence of P2O5 and KOH. The product (1) obtained in the form of white crystals (10.2 g, 90%) was free from nitride and hydrazine, it was characterized by IR and it was stored under vacuum in the presence of KOH. [0465] (1): m.p.: 203-205 C. (dec.). Rf: 0.26-0.29. [0466] IR (KBr) nu/cm-1: disappearance of the azide band (2105), 3428 (O-H), 2928 (C-H), 1080, 1029 (C-O-C). [0467] 1H-NMR (DMSO-d6): delta=5.78-5.63 (m, 14H), 4.90-4.85 (m, 7H), 4.50-4.45 (m, 6H), 3.66-3.54 (m, 28H), 3.42-3.24 (overlap with HDO, m, 16H). [0468] 13C-NMR (125 MHz, DMSO-d6): epsilon=101.8, 82.9, 81.6, 81.5, 73.0, 72.3, 72.1, 59.9. |
| 85% |
|
Compound 3 obtained in (ii) above, (24.1 g, 21 mmol) and Ph3P (27.55 g, 105 mmol) were dissolved in DMF (150 ml). The reaction mixture was stirred at 25 C. for 2 h. Concentrated ammonia solution (48.2 ml) was added and a white precipitate was formed. The reaction mixture was maintained at room temperature for 10 h. The precipitate was then removed by filtration and the solvent was evaporated under reduced pressure. The residue was added to ethanol (1500 ml). The solid precipitate was filtered, washed with ether (100 ml) and dried under high vacuum yielding 20.1 g (85% yield) of 4. TLC analysis of 4 performed on silica plates (EtOAc:2-propanol:conc. NH4OH: water-7:7:5:4) showed one major spot (Rf=0.20). 1H NMR (DMSO-d6) delta: 3.30-3.65 (m, 42H), 4.44-4.46 (m, 6H), 4.83, 4.89 (two d, 7H), 5.62-5.78 (m, 14H). HPLC (Luna 5u NH2 100 A, size 250-4.6 mm, mobile phase 65% acetonitrile-35% H2O, flow 1.2 ml/min), Rt=9.6 min. |






 Chemistry
Chemistry
 Pharmaceutical Intermediates
Pharmaceutical Intermediates
 Inhibitors/Agonists
Inhibitors/Agonists
 Material Science
Material Science







 For Research Only
For Research Only
 120K+ Compounds
120K+ Compounds
 Competitive Price
Competitive Price
 1-2 Day Shipping
1-2 Day Shipping