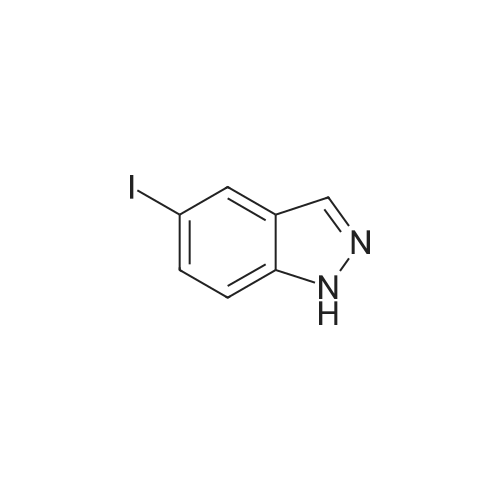
| 96% |
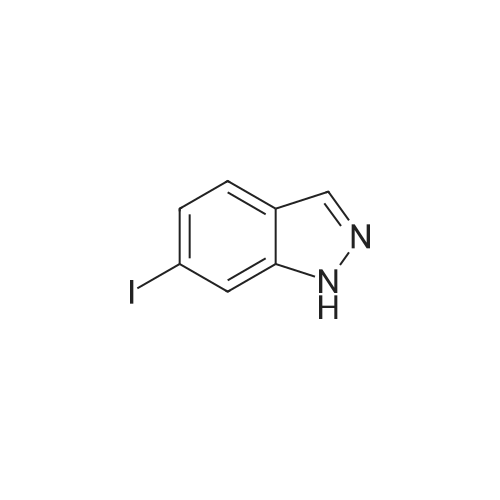
With tris-(dibenzylideneacetone)dipalladium(0); cesiumhydroxide monohydrate; 4,5-bis(diphenylphosphino)-9,9-dimethylxanthene; In N,N-dimethyl-formamide; at 100℃; for 4.5h;Inert atmosphere; |
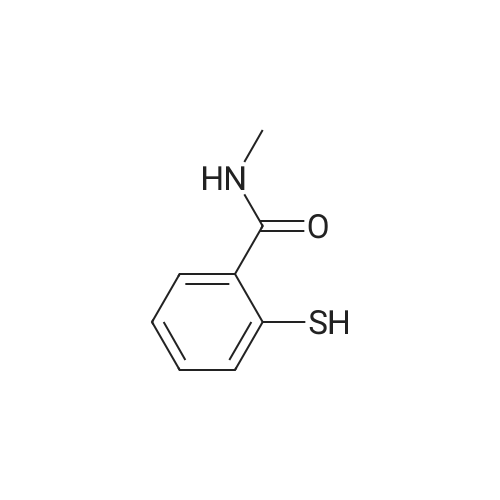
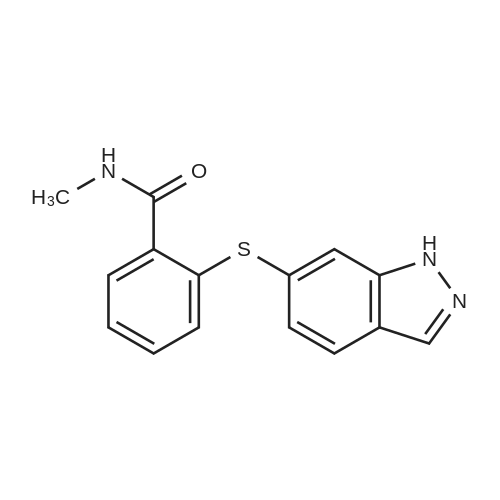
N,N-Dimethylformamide (25 mL) was added to 6-iodo-1H-indazole (5.51 g, 22 mmol), 2-mercapto-N-methylbenzamide(5.16 g, 31 mmol), Pd2(dba)3 (1.02 g, 1.1 mmol), 4,5-bis(diphenylphosphino)-9,9-dimethylxanthene (1.46 g,2.5 mmol) and cesium hydroxide monohydrate (5.67 g, 33 mmol) and stirred under an argon atmosphere at 100C for4.5 hours. The solvent was distilled away from the reaction solution under a reduced pressure, the residue was dissolvedin ethyl acetate and washed with water and saturated saline. The organic layer was dried with anhydrous sodium sulfate,and the solvent was distilled away under a reduced pressure. The residue was purified by silica gel column chromatography(hexane/ethyl acetate (v/v) = 1/3) to obtain the title compound (6.14 g, 96%) as a pale orange solid.1H NMR (400 MHz, DMSO-d6) δ 13.13 (1H, br s), 8.36 (1H, br q, J = 4.4 Hz), 8.10 (1H, s), 7.78 (1H, br d, J = 8.4 Hz),7.59 (1H, br s), 7.48-7.46 (1H, m), 7.29 (1H, td, J = 7.6, 1.6 Hz), 7.25 (1H, td, J = 7.6, 1.6 Hz), 7.08 (1H, dd, J = 8.4, 1.6Hz), 6.99-6.97 (1H, m), 2.76 (3H, d, J = 4.4 Hz) |
|
With cesium hydroxide; 4,5-bis(diphenylphosphino)-9,9-dimethylxanthene;tris-(dibenzylideneacetone)dipalladium(0); In 1-methyl-pyrrolidin-2-one; water; at 80℃; for 18.5h;Inert atmosphere; |
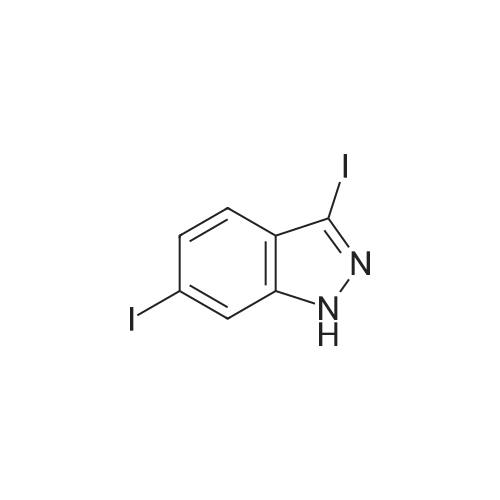
A 5 L three neck flask was equipped with a mechanical stirrer, a temperature probe, and a N2 inlet. The flask was charged with 6 -iodoindazole (200 g) followed by 2 - mercapto-N-methylbenzamide (144 g), Pd2(dba)3(3.75 g), Xantphos (4.74 g), NMP (1.2 L), and 50% aqueous CsOH solution (150 mL) in that order. Stirring was then commenced. The dark reaction mixture was degassed three times by alternately connecting to house vacuum and nitrogen. The mixture was heated to 80 C over a period of half an hour and maintained at the same temperature for 18 hours. The reaction was monitored by HPLC . It was noted that heating may be discontinued when the amount of 6-diiodoindazole is < 3%. The reaction mixture was allowed to cool to room temperature. An aqueous solution of Na HSO3 was prepared by adding 90 g of solid NaHSO3 into 1.5 L of deionized water with strong stirring. This solution was then set aside until the reaction quench step as described below. The reaction mixture in the 5 L flask was chilled in an ice-water bath until an internal temperature of 0.9 C was reached. KOH (183 g) was then charged in a single portion and the resulting mixture was allowed to stir for half an hour in ice-water bath (slight exotherm, highest point 4.0 C). Iodine (417 g) was dissolved in NMP (420 mL) in a separate flask with stirring. Once complete dissolution of iodine was been confirmed, the dark mixture was charged to a 1 L addition funnel. The iodine/NMP solution was then added dropwise over 1 h to the reaction mixture. (Note: the addition is exothermic and the internal reaction temperature must therefore be controlled via external cooling in addition to the controlled addition rate; the internal tempe rature should be kept between 0 C and 16.8C). Upon complete addition the final temperature was 14.5C. The flask was then taken out of the bath and the in ternal temperature reached 21.1C in 70 min. The mixture was allowed to stir at room temperature for three hours, at which time, analysis of an aliquot sample indicated the reacti on was complete (<3% left). Upon confirmation of reaction completion (HPLC), the flask was re -immersed in the icewater bath. The aqueous NaHSO3 solution prepared as described previously was added slowly over 40 minutes from an addition funnel. ( Note: this addition is exothermic and the internal reaction temperature must therefore be controlled via external cooling in addition to the controlled addition rate; the internal temper ature should be kept below15.7 C). Upon complete addition the reaction was a slurry of light yellow solids. The mixture was allowed to stir at ambient temperature overnight. The solid produ ct was collected by filtration. The wet cake was recharged back into the 5 L flask and the funnel was rinsed with 1.5 L of water, and the r inses were also charged into the 5 L flask. The mixture was stirred for one hour and filtered. The wet cake was recharged back to the 5 L flask, and the funnel was rinsed with 1.5 L of methanol, and the rinses were also charged into the 5 L flask. The mixture was heated at 45 C for two hours, then allowed to cool. The mixture was filtered and the cake was washed with 500 mL of MeOH, and sucked dry. The product (cake) was placed in a vacuum oven at 60 C for 18 h to afford 317 g of 2-(3-lodo-1H-indazol-6-ylsulfanyl)-N-methyl-benzamide. |
|
With cesium hydroxide;tris-(dibenzylideneacetone)dipalladium(0); 4,5-bis(diphenylphosphino)-9,9-dimethylxanthene; In 1-methyl-pyrrolidin-2-one; water; at 80℃; for 18.5h; |
A 5 L three neck flask was equipped with a mechanical stirrer, a temperature probe, and a N2 inlet. The flask was charged with <strong>[261953-36-0]6-iodoindazole</strong> (200 g) followed by 2-mercapto-N-methylbenzamide (144 g), Pd2(dba)3 (3.75 g), Xantphos (4.74 g), NMP (1.2 L), and 50% aqueous CsOH solution (150 mL) in that order. Stirring was then commenced. The dark reaction mixture was degassed three times by alternately connecting to house vacuum and nitrogen. The mixture was heated to 80 C. over a period of half an hour and maintained at the same temperature for 18 hours. The reaction was monitored by HPLC. It was noted that heating may be discontinued when the amount of 6-diiodoindazole is <3%. The reaction mixture was allowed to cool to room temperature. An aqueous solution of NaHSO3 was prepared by adding 90 g of solid NaHSO3 into 1.5 L of deionized water with strong stirring. This solution was then set aside until the reaction quench step as described below. The reaction mixture in the 5 L flask was chilled in an ice-water bath until an internal temperature of 0.9 C. was reached. KOH (183 g) was then charged in a single portion and the resulting mixture was allowed to stir for half an hour in ice-water bath (slight exotherm, highest point 4.0 C.). Iodine (417 g) was dissolved in NMP (420 mL) in a separate flask with stirring. Once complete dissolution of iodine was been confirmed, the dark mixture was charged to a 1 L addition funnel. The iodine/NMP solution was then added dropwise over 1 h to the reaction mixture. (Note: the addition is exothermic and the internal reaction temperature must therefore be controlled via external cooling in addition to the controlled addition rate; the internal temperature should be kept between 0 C. and 16.8 C.). Upon complete addition the final temperature was 14.5 C. The flask was then taken out of the bath and the internal temperature reached 21.1 C. in 70 min. The mixture was allowed to stir at room temperature for three hours, at which time, analysis of an aliquot sample indicated the reaction was complete (<3% left). Upon confirmation of reaction completion (HPLC), the flask was re-immersed in the ice-water bath. The aqueous NaHSO3 solution prepared as described previously was added slowly over 40 minutes from an addition funnel. (Note: this addition is exothermic and the internal reaction temperature must therefore be controlled via external cooling in addition to the controlled addition rate; the internal temperature should be kept below 15.7 C.). Upon complete addition the reaction was a slurry of light yellow solids. The mixture was allowed to stir at ambient temperature overnight. The solid product was collected by filtration. The wet cake was recharged back into the 5 L flask and the funnel was rinsed with 1.5 L of water, and the rinses were also charged into the 5 L flask. The mixture was stirred for one hour and filtered. The wet cake was recharged back to the 5 L flask, and the funnel was rinsed with 1.5 L of methanol, and the rinses were also charged into the 5 L flask. The mixture was heated at 45 C. for two hours, then allowed to cool. The mixture was filtered and the cake was washed with 500 mL of MeOH, and sucked dry. The product (cake) was placed in a vacuum oven at 60 C. for 18 h to afford 317 g of 2-(3-Iodo-1H-indazol-6-ylsulfanyl)-N-methyl-benzamide. |






 Chemistry
Chemistry
 Pharmaceutical Intermediates
Pharmaceutical Intermediates
 Inhibitors/Agonists
Inhibitors/Agonists
 Material Science
Material Science







 For Research Only
For Research Only
 120K+ Compounds
120K+ Compounds
 Competitive Price
Competitive Price
 1-2 Day Shipping
1-2 Day Shipping