* All experimental methods are cited from the reference, please refer to the original source for details. We do not guarantee the accuracy of the content in the reference.
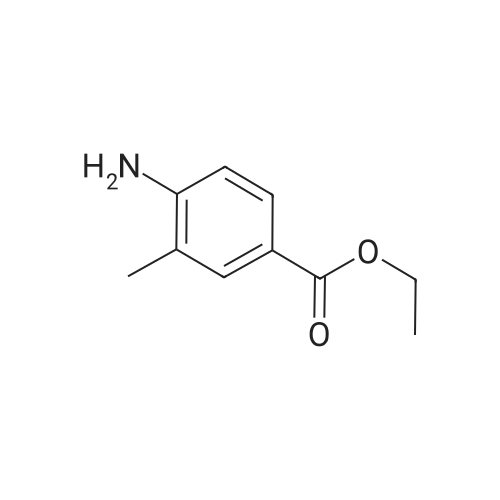
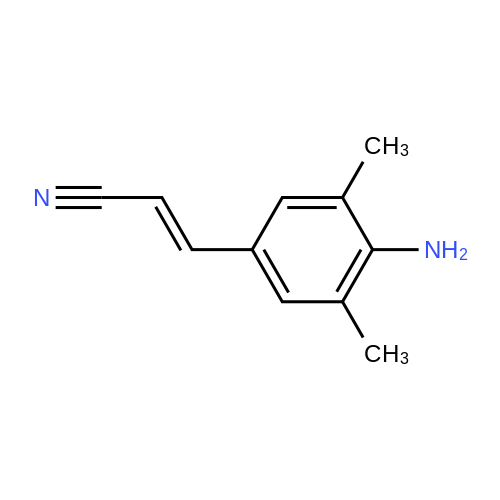
(2E)-3-(4-amino-3,5-dimethylphenyl)acrylonitrile hydrochloride[ No CAS ]
[ 244768-32-9 ]
[ 500287-72-9 ]
Yield
Reaction Conditions
Operation in experiment
89.6%
b) A mixture of 93.9 g (0. 45 mol) of the hydrochloric acid salt of intermediate [(II),] prepared according to Example A2, and 103.8 g (0.45 mol) of intermediate (III-a) in 0.9 1 of acetonitrile was prepared under nitrogen atmosphere. The mixture was stirred and refluxed for 24 hours, then allowed to cool to 50 C. A solution of K2C03 (124.4 g, 0.9 mol) in H20 (0.45 1) was added over a period of 15-20 minutes at 40-50 C, followed by stirring for 1 hour at 50 C. The precipitate was separated and washed twice with 0.045 1 of acetonitrile, followed by drying at 50C under reduced pressure. 73.3 g of the obtained solid and 400 ml of EtOH were mixed and refluxed for 2 hours, then allowed to cool to room temperature. The precipitate was filtered and the residue was washed with 50 ml of EtOH. The obtained residue was dried overnight at 50C under reduced pressure. Yield: 65.7 g (89. 6 %) of 4-[[4-[[4-(2-cyanoethenyl)-2, 6- dimethylphenyl] amino]-2-pyrimidinyl] amino] benzonitrile (E) (Compound X).
68.6%
A mixture of 93.9 g (0.45 mol) of the hydrochloric acid salt of intermediate (II), prepared according to Example A2, and 109 g (0.4725 mol) of intermediate (III-a) in 1.81 of of acetonitrile was prepared under nitrogen atmosphere. The mixture was stirred and refluxed for 69 hours, then allowed to cool to 55 C. The mixture was filtered and the residue was washed with 200 ml of acetonitrile, followed by drying under reduced pressure at 50C overnight. 144, 6 g (0.3666 mol) of the obtained solid was brought in 1 l of K2CO3 10% aqueous solution. The mixture was stirred at room temperature followed by filtration. The obtained residue was washed twice with water followed by drying at 50C under reduced pressure. The residue was brought in 6.55 1 isopropanol and the mixture was refluxed, then stirred overnight and filtered at room temperature. The residue was dried at 50C under reduced pressure. Yield: 113.2 g [(68.] 6 %) of 4-[[4-[[4-(2-cyanoethenyl)-2,6-dimethylphenyl]amino-2-pyrimidinyl]amino]benzonitrile (E) (Compound X).
With toluene-4-sulfonic acid; In 1,4-dioxane; at 100 - 110℃; for 14h;Product distribution / selectivity;
Example 8:Preparation of RilpivirineTo a mixture of 4-(4-chloropyrimidin-2-ylamino)benzonitrile (4.5 gm) as obtained in example 3, (E)-3-(4-amino-3,5-dimethylphenyl)acrylonitrile hydrochloride (4.07 gm) as obtained in example 7 and p-toluenesulfonic acid monohydrate (4.45 gm) was added 1,4-dioxane (90 ml) under stirring. The mixture was then heated to 100 to 110C and stirred for 14 hours. The solution was then cooled to room temperature and then added saturated sodium bicarbonate solution. The layers were separated and the aqueous layer was extracted with ethyl acetate. The combined organic layers were dried with sodium sulfate and then concentrated to obtain a crude solid.The crude solid obtained above was dissolved in acetone and stirred for 1 hour at room temperature. The separated solid was filtered and then dried to obtain 4 gm of rilpivirine.
135 g
In 1-methyl-pyrrolidin-2-one; at 85 - 95℃; for 16h;
To a mixture of 4-(4-chloropyrimidin-2-ylamino)benzonitrile (85 gm) as obtained in preparative example 1 and (E)-3-(4-amino-3,5-dimethylphenyl)acrylonitrile hydrochloride (69 gm) as obtained in preparative example 2 was added N- methylpyrrolidone (425 ml) under stirring. The mixture was then heated to 85 to 95C and stirred for 16 hours. The solution was then cooled to room temperature and then added water (1 105 ml). The reaction mass was stirred for 1 hour 30 minutes at room temperature and filtered. The solid obtained was then dried to obtain 135 gm of rilpivirine as a white solid.Chromatographic purity of rilpivirine: 98.6%;Content of Z-isomer: 1.2%.
135 g
In 1-methyl-pyrrolidin-2-one; at 85 - 95℃; for 16h;
Example 1 Preparation of rilpivirine To a mixture of 4-(4-chloropyrimidin-2-ylamino)benzonitrile (85 gm) as obtained in preparative example 1 and (E)-3-(4-amino-3,5-dimethylphenyl)acrylonitrile hydrochloride (69 gm) as obtained in preparative example 2 was added N-methylpyrrolidone (425 ml) under stirring. The mixture was then heated to 85 to 95 C. and stirred for 16 hours. The solution was then cooled to room temperature and then added water (1105 ml). The reaction mass was stirred for 1 hour 30 minutes at room temperature and filtered. The solid obtained was then dried to obtain 135 gm of rilpivirine as a white solid.
With sodium hydride; In N,N-dimethyl-formamide; mineral oil; at -20 - 20℃;Inert atmosphere;
General procedure: A solution of 14a-c (2 mmol), appropriate aryl acetonitrile (3 mmol) and anhydrous DMF (30 mL) was stirred for 0.5 h at -20 C. Then NaH (0.14 g, 3.5 mmol; 60% dispersion in mineral oil) was added portionwise at -20 C under Ar. The mixture was stirred at RT for 48-72 h under Ar, and then stirred under air for another 36-72 h at RT. The resulting mixture was poured into water and extracted with EtOAc. The combined organic layers were dried by Na2SO4, filtered and concentrated in vacuo. Purification by flash chromatography (silica gel; EtOAc/petroleum ether, 1:6 to 1:4) gave 16a-z.
(E)-3-(4-amino-3,5-dimethylphenyl)-2-acrylonitrile hydrochloride[ No CAS ]
[ 244768-32-9 ]
[ 500287-72-9 ]
Yield
Reaction Conditions
Operation in experiment
58%
(E)-3-(4-Amino-3,5-dimethylphenyl)acrylonitrile hydrochloride (48 g, 0.23 mol) and 4-(4-chloropyrimidin-2-ylamino)benzonitrile (55.7 g, 0.24 mol, 1.05 eq) were mixed with acetonitrile (480 mL, 10V) at 25C affording a yellow suspension. The suspension was stirred and heated to reflux (82C). Stirring was continued at the same temperature for 48 h, and the suspension was then cooled to 50C over a period of 0.5 h. A solution of potassium carbonate (63.5 g, 0.46 mol, 2 eq) in water (240 mL) was added dropwise over 15 min at 50C, and stirring was continued for another 1 h. The resulting solid was isolated by vacuum filtration, washed with acetonitrile/water (40+30 ml) and air-dried on the filter. The air-dried filter cake (74.2 g) was mixed with EtOH (abs) (410 mL, 5.5V vs. crude Rilpivirine), and the resulting slurry was heated to reflux (78C) with stirring. The hot suspension was stirred for 2 h at 78C, and then cooled to 25C over a period 0.5 h. The resulting solid was isolated by vacuum filtration; the filter cake was washed with EtOH (abs) (75 ml) and dried in a vacuum oven (10 mbar) at 50C for 16 h to afford Rilpivirine base form II (48.9 g, 58.1% yield, 98.4% purity) as a yellow solid.
4 g
With toluene-4-sulfonic acid; In 1,4-dioxane; water; at 100 - 110℃; for 14h;
Example 8 Preparation of Rilpivirine [0088] To a mixture of 4-(4-chloropyrimidin-2-ylamino)benzonitrile (4.5 gm) as obtained in example 3, (E)-3-(4-amino-3,5-dimethylphenyl)acrylonitrile hydrochloride (4.07 gm) as obtained in example 7 and p-toluenesulfonic acid monohydrate (4.45 gm) was added 1,4-dioxane (90 ml) under stirring. The mixture was then heated to 100 to 110 C. and stirred for 14 hours. The solution was then cooled to room temperature and then added saturated sodium bicarbonate solution. The layers were separated and the aqueous layer was extracted with ethyl acetate. The combined organic layers were dried with sodium sulfate and then concentrated to obtain a crude solid. [0089] The crude solid obtained above was dissolved in acetone and stirred for 1 hour at room temperature. The separated solid was filtered and then dried to obtain 4 gm of rilpivirine.
In acetonitrile; for 69h;Inert atmosphere; Reflux;
Compound 1 was also prepared as follows: (0510) A mixture of 93.9 g (0.45 mol) of the hydrochloric acid salt of intermediate 3, prepared according to Example A1c), and 109 g (0.4725 mol) of intermediate 5 in 1.81 of acetonitrile was prepared under nitrogen atmosphere. The mixture was stirred and refluxed for 69 hours, then allowed to cool to 55 C. The mixture was filtered and the residue was washed with 200 ml of acetonitrile, followed by drying under reduced pressure at 50 C. overnight. 144.6 g (0.3666 mol) of the obtained solid was brought in 11 of K2CO3 10% aqueous solution. The mixture was stirred at room temperature followed by filtration. The obtained residue was washed twice with water followed by drying at 50 C. under reduced pressure. The residue was brought in 6.551 isopropanol and the mixture was refluxed, then stirred overnight and filtered at room temperature. The residue was was dried at 50 C. under reduced pressure. Yield: 113.2 g (68.6%) of 4-[[4-[[4-(2-cyanoethenyl)-2,6-dimethylphenyl]amino]-2-pyrimidinyl]amino]benzonitrile (E) (compound 1).
A mixture of intermediate 3 (0.034 mol) and intermediate 5 (0.0174 mol) was stirred at 150 C. for 1 hour and taken up in K2CO3 10%/CH2Cl2/CH3OH. The organic layer was separated, dried (MgSO4), filtered, and the solvent was evaporated. The residue (10 g) was purified by column chromatography over silica gel (eluent: CH2Cl2/ethyl acetate 80/20; 15-40 mum). Fraction 1 was crystallized from iPrOH. The precipitate was filtered off and dried. Yield: 1.3 g of 4-[[4-[[4-(2-cyanoethenyl)-2,6-dimethylphenyl]amino]-2-pyrimidinyl]amino]benzonitrile (E) (compound 1) (20%).
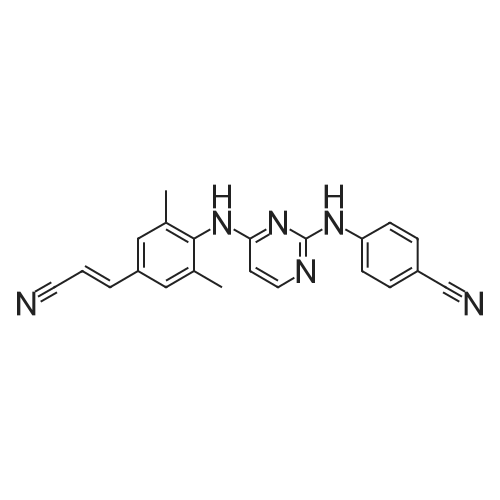
4-[[4-[[4-(2-cyanoethenyl)-2,6-dimethylphenyl]amino]-2-pyrimidinyl]amino]benzonitrile[ No CAS ]
[ 500287-72-9 ]
Yield
Reaction Conditions
Operation in experiment
8.7%
In 1-methyl-pyrrolidin-2-one; at 95℃; for 17h;Molecular sieve;
NMP (80 ml; 0.09% of water, dried 4A MS - molecular sieves) is added to a mixture of 8.35 g (40 mmol) of 1.HCI and 10.84 g (47 mmol) of 2 and the mixture is heated upto 95C NMP (80 ml; 0.09 % of water, dried 4A MS - molecular sieves) is added for17 h. A solution of sodium carbonate (50 g of Na2CO3, topped up with water to 500ml) is gradually added to the mixture cooled down to 30C until pH 7.5-8 is obtained(58 ml in total). Then, water (200 ml) is gradually added and the suspension is stirredat the room temperature (it) for 3 h. The solid substance is aspirated on glass frit,washed with aqueous DMF (1:2 vol./vol.; 30 ml) and dried. 15.13 g (103.2%) of thecrude product is obtained; UPLC 72.5%, Z-isomer 8.7%, hydroxy derivative 13.3%.
3-(4-amino-3,5-dimethylphenyl)acrylonitrile[ No CAS ]
[ 244768-32-9 ]
4-[[4-[[4-(2-cyanoethenyl)-2,6-dimethylphenyl]amino]-2-pyrimidinyl]amino]benzonitrile[ No CAS ]
[ 500287-72-9 ]
Yield
Reaction Conditions
Operation in experiment
In dichloromethane; at 160 - 165℃; for 1h;
The aniline derivative I (hydrochloride 710 mg, 3.38 mmol) is dosed into a flask (50ml), DCM (dichloromethane) (10 ml) and a 10% aqueous solution of potassiumcarbonate (5 ml) are added. The suspension slowly passes into a solution while being stirred at it. The organic phase is separated, dried and evaporated until dry at a reduced pressure. The base of the aniline derivative 1 (582 mg, 99.32%) is obtained. Chloropyrimidine 2 (401 mg, 1.7 mmol) is added to it and the mixture is melted in abath at a temperature of 160-165C for 1 hour. After cooling, a 10% aqueous solution of K2C03 (5 ml), as well as a solvent mixture of DCM and methanol (2:1, 10 ml) are added to the mixture. The organic layer is separated, evaporated and the obtained evaporation product (0.9 g) is purified by column chromatography, eluent DCM/ethylacetate 80:20. The product is obtained in the 51.8% yield (330 mg), HPLC 84.18%, Z25 isomer 14.8%.






 Chemistry
Chemistry
 Pharmaceutical Intermediates
Pharmaceutical Intermediates
 Inhibitors/Agonists
Inhibitors/Agonists
 Material Science
Material Science







 For Research Only
For Research Only
 120K+ Compounds
120K+ Compounds
 Competitive Price
Competitive Price
 1-2 Day Shipping
1-2 Day Shipping