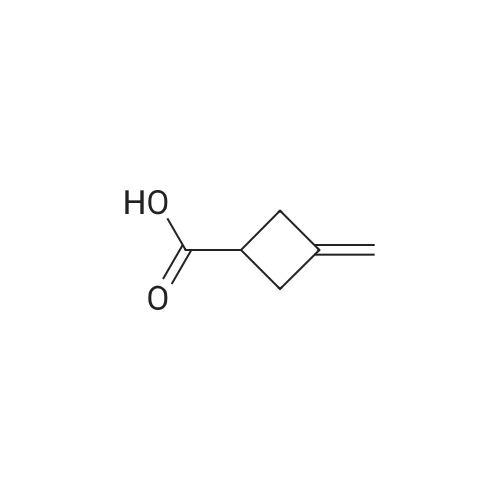
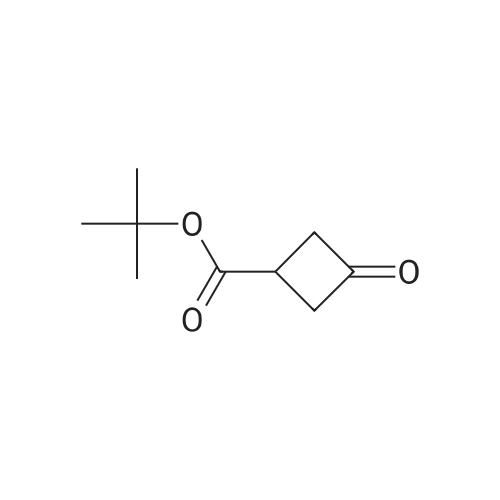
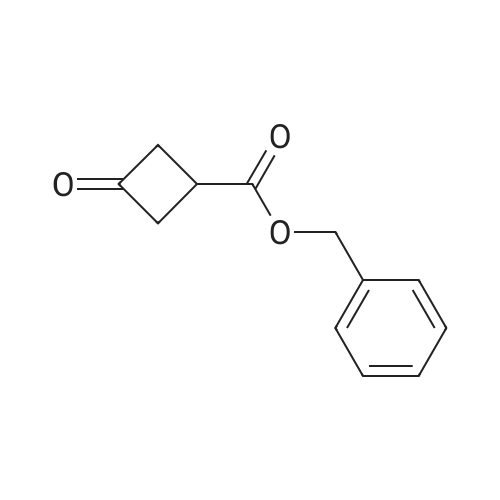
| 41.9% |
With potassium carbonate; In acetone; for 10h;Reflux; |
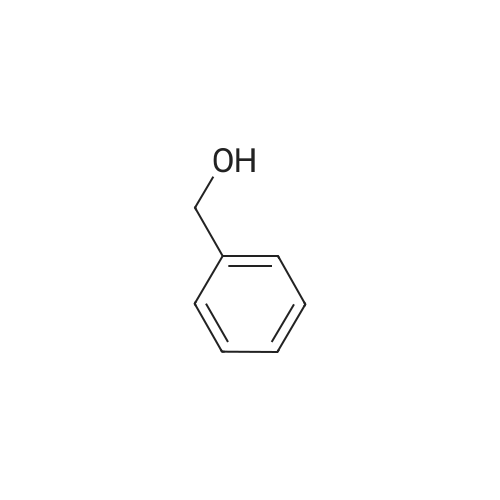
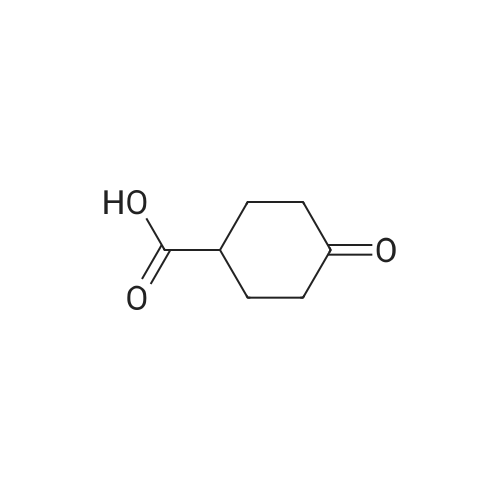
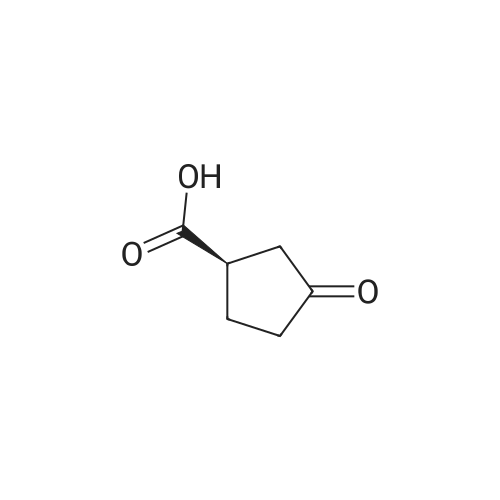
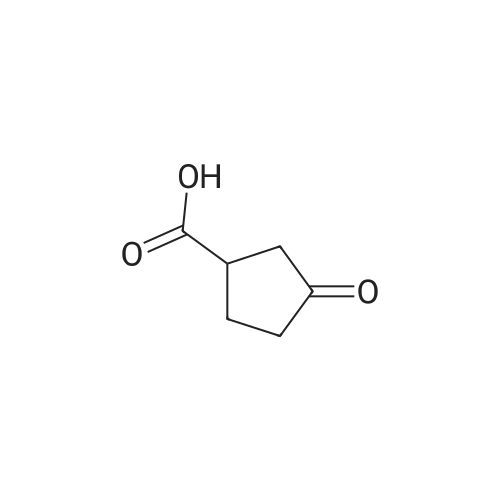
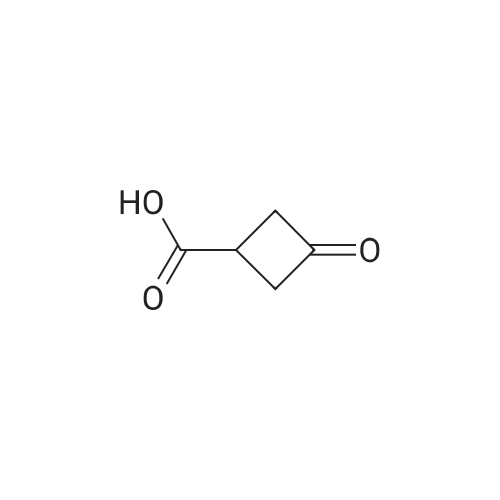
3-Oxocyclobutanecarboxylic acid(3.0 g, 26.3 mmol), benzyl bromide(6.7 g, 39.4 mmol) and potassium carbonate(7.3 g, 52.6 mmol) was dissolved in acetone(30 mL), and then heated to reflux for 10 hours. After TLC showed that the reaction was completed, the reaction mixture was concentrated under reduce pressure to remove the solvent, added water (20 mL), and extracted with ethyl acetate (150 mL*2). The combined organic phase was washed with saturated salt water(50 mL), dried with anhydrous sodium sulfate, filtered and concentrated under vacuum. The residue was purified through silica gel column chromatography (petroleum ether/ethyl acetate=10:1) to give benzyl 3-oxocyclobutane-carboxylate (2.5 g, 41.9% yield) as a colorless liquid. MS (ESI) Calcd. for C12H12O3 [M+H]+ 205, Found 205. |
| 36.7% |
With caesium carbonate; In acetonitrile; at 20℃; for 20h;Inert atmosphere; |
To a mixture of commercially available 3-oxocyclobutanecarboxylic acid (13.50 g,118.32 mmol) and Cs2003 (46.26 g, 141.98 mmol) in CH3CN (120 mL), was added bromomethylbenzene (21 .25 g, 124.24 mmol) in one portion at r.t. under N2. The mixture was stirred at r.t. for 20 hours. LCMS and TLC showed the reaction was completed. The residue was poured into water and stirred for 20 mm. The aqueous phase was extracted with EtOAc (120 mL). Thecombined organic phase was washed with saturated brine (140 mL), dried with anhydrous Na2SO4, filtered and concentrated in vacuum. The residue was purified by silica gel chromatography to afford reagent KR-Si (8.86 g, 36.7%). |
|
With triethylamine; In tetrahydrofuran; at 20℃; for 2h; |
Triethylamine (2.0 mL) and benzyl bromide (1.2 mL) were added to a solution of 3-oxocyclobutanecarboxylic acid (J. Org. Chem., vol. 53, pp. 3841-3843 (1981)) (995 mg) in tetrahydrofuran (5.0 mL), and the mixture was stirred at room temperature for 2 hours. The reaction mixture was diluted with ethyl acetate, and was sequentially washed with 1N aqueous HCl, saturated aqueous sodium hydrogencarbonate, and saturated brine, followed by drying over sodium sulfate anhydrate. The solvent was distilled away under reduced pressure, and the residue was purified by silica gel column chromatography (ethyl acetate : hexane = 1:6), to thereby give the title compound (886 mg).1H-NMR(CDCl3) delta:3.22-3.33(3H, m), 3.37-3.48(2H, m), 5.19(2H, s), 7.31-7.42(5H, m).MS(FAB)m/z:205(M+H+). |
|
With sodium hydrogencarbonate; triethylamine; In tetrahydrofuran; |
REFERENTIAL EXAMPLE 150 Benzyl 3-oxocyclobutanecarboxylate: Triethylamine (2.0 ml) and benzyl bromide (1.2 ml) were added to a solution of 3-oxocyclobutanecarboxylic acid (J. Org. Chem., Vol. 53, pp. 3841-3843, 1981) (995 mg) in tetrahydrofuran (5.0 ml), and the mixture was stirred at room temperature for 2 hours. The reaction mixture was diluted with ethyl acetate, and washed successively with 1N hydrochloric acid, a saturated aqueous solution of sodium hydrogencarbonate and saturated saline and dried over anhydrous sodium sulfate. The solvent was then distilled off under reduced pressure, and the resultant residue was purified by column chromatography on silica gel (ethyl acetate:hexane=1:6) to obtain the title compound (886 mg). 1H-NMR (CDCl3) delta: 3.22-3.33(3H,m), 3.37-3.48(2H,m), 5.19(2H,s), 7.31-7.42(5H,m). |
|
|
Step A:3-Oxocyclobutanecarboxylic acid (2-A) (1.0 g, 10.0 mmol) was dissolved in anhydrous ethanol (25 ml), and cesium carbonate (1.66 g, 5.1 mmol) was added. After stirring at room temperature under nitrogen for 4 hours, the reaction mixture was concentrated. The residue was redissolved in anhydrous acetonitrile (50 ml) and treated with benzyl bromide (1.2 ml, 10.0 ml). The mixture was allowed to stir at room temperature under nitrogen for 12 hours. Solvent was then removed under reduced pressure and the residue was partitioned between ethyl acetate and water. The crude product was purified with silica gel chromatography eluting with a gradient of 100% hexane to 96% hexane/ethyl acetate to give 2-B. |
|
With triethylamine; In tetrahydrofuran; at 20℃; for 2h; |
[Referential Example 150] benzyl 3-oxocyclobutanecarboxylate: triethylamine (2.0 ml) and benzyl bromide (1.2 ml) were added to a solution of 3-oxocyclobutanecarboxylic acid (J. Org. Chem., Vol. 53, pp. 3841-3843, 1981) (995 mg) in tetrahydrofuran (5.0 ml), and the mixture was stirred at room temperature for 2 hours.. The reaction mixture was diluted with ethyl acetate, and washed successively with 1N hydrochloric acid, a saturated aqueous solution of sodium hydrogencarbonate and saturated saline and dried over anhydrous sodium sulfate.. The solvent was then distilled off under reduced pressure, and the resultant residue was purified by column chromatography on silica gel (ethyl acetate:hexane = 1:6) to obtain the title compound (886 mg).1H-NMR (CDCl3) delta: 3.22-3.33(3H,m), 3.37-3.48(2H,m), 5.19(2H,s), 7.31-7.42(5H,m). MS (FAB) m/z: 205(M+H+). |
|
With potassium carbonate; In acetonitrile; at 20℃; for 24h; |
BnBr was added to the mixture of compound 1 and K2CO3 in acetonitrile 150 mL. The mixture was stirred at room temperature over 24 h and the solid was filtered. The solvent was removed and the residue was purified by column chromatography to give compound 2. |
|
With potassium carbonate; In acetonitrile; at 20℃; for 24h; |
Step 1: benzyl 3-oxocyclobutanecarboxylate (2) Referring to Scheme 6A, BnBr was added to the mixture of compound 1 and K2CO3 in acetonitrile 150 mL. The mixture was stirred at room temperature over 24 h and the solid was filtered. The solvent was removed and the residue was purified by column chromatography to give compound 2. |
|
With potassium carbonate; In [(2)H6]acetone; hexane; |
Step A: Benzyl 3-oxocyclobutanecarboxylate A mixture of 3-oxocyclobutanecarboxylic acid (5 g, 44 mmol), potassium carbonate (12 g, 88 mmol) and benzyl bromide (11.2 g, 66 mmol) in acetone (50 mL) was refluxed for 16 h. The solvent was then removed under reduced pressure and the residue was partitioned between ethyl acetate and water. Combined organic layers were dried over anhydrous MgSO4, filtered and concentrated. The residue was purified with silica gel chromatography eluting with a gradient of 100% hexane to 96% hexane/EtOAc to give the desired compound. 1H NMR (400 MHz, CDCl3): delta 7.45-7.27 (m, 5H), 5.19 (s, 2H), 3.55-3.36 (m, 2H), 3.33-3.11 (m, 3H). |
|
With potassium carbonate; In acetone; for 16h;Reflux; |
Method Step A: Benzyl 3-oxocyclobutanecarboxylate. A mixture of 3-oxocyclobutanecarboxylic acid (5 g, 44 mmol), potassium carbonate (12 g, 88 mmol) and benzyl bromide (1 1.2 g, 66 mmol) in acetone (50 mL) was refluxed for 16 h. The solvent was then removed under reduced pressure and the residue was partitioned between ethyl acetate and water. Combined organic layers were dried over anhydrous MgS04, filtered and concentrated. The residue was purified with silica gel chromatography eluting with a gradient of 100% hexane to 96% hexane / EtOAc to give the desired compound. FontWeight="Bold" FontSize="10" H NMR (400 MHz, CDC13): delta 7.45 - 7.27 (m, 5H), 5.19 (s, 2H), 3.55 - 3.36 (m, 2H), 3.33 - 3.11 (m, 3H). |
|
|
To a solution of commercially available 3-oxocyclobutanecarboxylic acid (14.9 g) in DMF (210 mL) were added potassium carbonate (27.07 g) and benzyl bromide (18.6 mL), and the mixture was stirred at room temperature for 2 hr. To the reaction mixture was added acetic acid (22.4 mL) over 12 min, and the mixture was stirred at room temperature for 10 min. Water (350 mL) was added, and the mixture was extracted 3 times with ethyl acetate. The organic layer was washed 4 times with saturated brine, dried over magnesium sulfate and concentrated. [1411] Toluene was added and the mixture was concentrated to give a crude product of compound 3-1. The obtained crude product of compound 3-1 was directly used in the next step. |
|
With potassium carbonate; In acetone; for 16h;Reflux; |
A mixture of 3-oxocyclobutanecarboxylic acid (5g, 44 mmol), potassium carbonate (12 g, 88 mmol) and benzyl bromide (11.2 g, 66 mmol) in acetone (50 mL) was refluxed for 16 h. The solvent was then removed under reduced pressure and the residue was partitioned between ethyl acetate and water. Combined organic layers were driedover anhydrous MgSO4, filtered and concentrated. The residue was purified with silica gel chromatography eluting with a gradient of 100% hexane to 96% hexane EtOAc to givethedesired compound. ?H NMR (400 MHz, CDC13): 7.45 - 7.27 (m, 511), 5.19 (s, 211), 3.55-3.36 (m, 211), 3.33 -3.11 (m, 311). |
|
With potassium carbonate; In acetone; for 16h;Reflux; |
Step A: Benzyl 3-oxocyclobutanecarboxylate. A mixture of 3-oxocycIobutanecarboxylic acid (5 g, 44 mmol), potassium carbonate ( 12 g, 88 mmol) and benzyl bromide (1 1.2 g, 66 mmol) in acetone (50 mL) was refluxed for 1 h. The solvent was then removed under reduced pressure and the residue was partitioned between ethyl acetate and water. Combined organic layers were dried over anhydrous MgSC , filtered and concentrated. The residue was purified with silica gel chromatography eluting with a gradient of 100% hexane to 96% hexane / EtOAc to give the desired compound. NMR (400 MHz, CDC13): delta 7.45 - 7.27 (m, 5H), 5.19 (s, 2H), 3.55 - 3.36 (m, 2H), 3.33 - 3.1 1 (m, 3H). |






 Chemistry
Chemistry
 Pharmaceutical Intermediates
Pharmaceutical Intermediates
 Inhibitors/Agonists
Inhibitors/Agonists
 Material Science
Material Science







 For Research Only
For Research Only
 120K+ Compounds
120K+ Compounds
 Competitive Price
Competitive Price
 1-2 Day Shipping
1-2 Day Shipping


-4-Amino-5-hydroxybicyclo[3.1.1]heptane-2-carboxylic Acid via an Asymmetric Intramolecular Mannich Reaction.png)