| 36% |
|
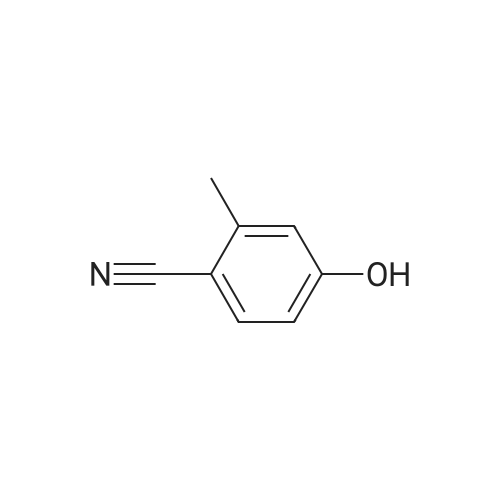
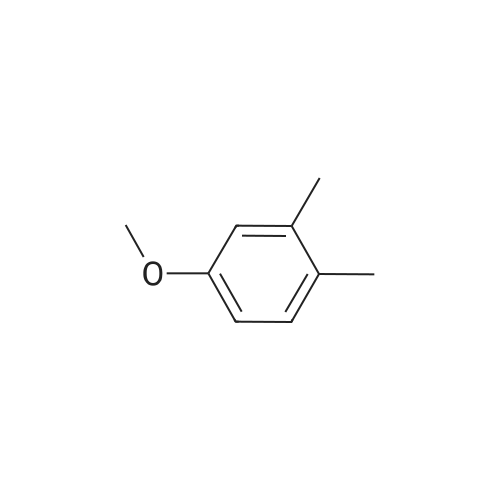
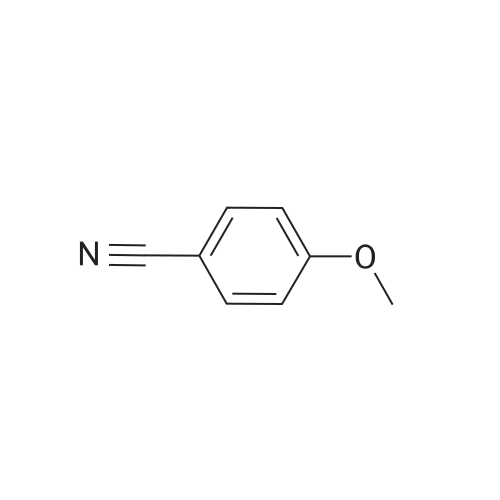
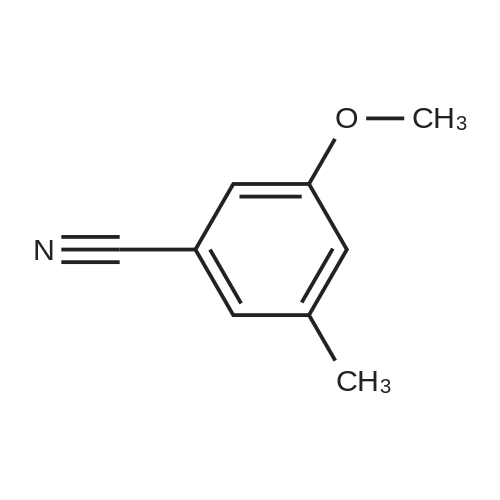
Preparation 42 4-Hydroxy-2-methyl benzonitrile Boron trichloride (1M in dichloromethane, 747 ml, 747 mmol) was added dropwise, at -78 C., to a suspension of commercially available <strong>[21883-13-6]4-methoxy-2-methyl-benzonitrile</strong> (44 g, 298 mmol) and tetrabutylammonium iodide (121 g, 327 mmol) in dichloromethane (750 ml), under nitrogen, over 40 minutes. Once the addition was complete, the yellow solution was warmed to room temperature and stirred for 16 hours at room temperature. The reaction mixture was then quenched by dropwise addition of water maintaining the internal temperature below 10 C. The mixture was filtered through Arbocel and the layers were separated. The aqueous layers were extracted again with dichloromethane (250 ml). The organic layers were combined, washed with a sodium thiosulphate solution (150 ml), dried over magnesium sulphate, filtered and concentrated under reduced pressure to give thick yellow oil. Trituration of the oil in dichloromethane, followed by filtration, provided a first crop of the title compound (10.8 g, 27%) as a white solid. The filtrate was evaporated and purified by flash chromatography on silica gel, eluding with pentane:ethyl acetate (70:30, by volume) to provide more of the title compound as a white solid (14.4 g, 36%). 1H-NMR (400 MHz, CDCl3): delta=2.46 (s, 3H), 6.68 (d, 1H), 6.72 (s, 1H), 7.45 (d, 1H); LRMS: APCl-: m/z 132 [M-H]-. |
|
|
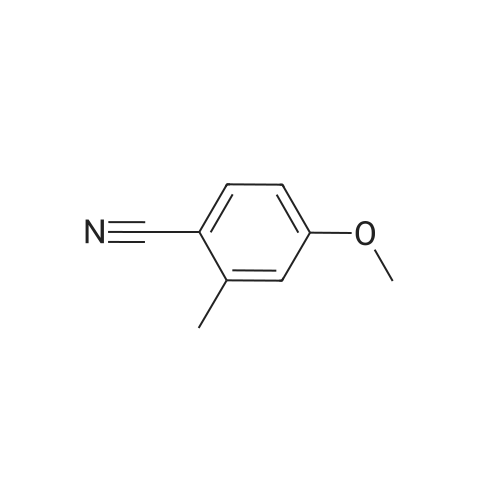
4-Hydroxy-2-methyl-benzonitrile A solution of <strong>[21883-13-6]4-methoxy-2-methylbenzonitrile</strong> (5.0 g, 33.97 mmol) in DCM (150 mL) is cooled down to 0 C. before adding dropwise a 1M BBr3 in DCM solution (136 mL, 136 mmol). The reaction mixture is allowed to reach rt and stirring is then continued at 45 C. for 5 days. Ice water (500 mL) is then added and the reaction mixture is stirred for 1 h before sat. aq. NaHCO3 (250 mL) is added. The mixture is extracted with DCM (200 mL then 4*100 mL) and the combined org. extracts are dried over MgSO4, filtered and evaporated to give the title compound as a brown solid (4.7 g); LC-MS: tR=0.76 min. 1H NMR (D6-DMSO): delta 2.38 (s, 3H), 6.73 (dd, J=8.5, 2.0 Hz, 1H), 6.79 (d, J=2.0 Hz, 1H), 7.55 (d, J=8.5 Hz, 1H), 10.49 (s, 1H). |
|
|
A solution of <strong>[21883-13-6]4-methoxy-2-methylbenzonitrile</strong> (5.0 g, 33.97 mmol) in DCM (150 mL) is cooled down to 0 C. before adding dropwise a 1M BBr3 in DCM solution (136 mL, 136 mmol). The reaction mixture is allowed to reach rt and stirring is then continued at 45 C. for 5 days. Ice water (500 mL) is then added and the reaction mixture is stirred for 1 h before sat. aq. NaHCO3 (250 mL) is added. The mixture is extracted with DCM (200 mL then 4×100 mL) and the combined organic extracts are dried over MgSO4, filtered and evaporated to give the title compound as a brown solid (4.7 g); LC-MS: tR=0.76 min. 1H NMR (D6-DMSO): delta 2.38 (s, 3H), 6.73 (dd, J=8.5, 2.0 Hz, 1H), 6.79 (d, J=2.0 Hz, 1H), 7.55 (d, J=8.5 Hz, 1H), 10.49 (s, 1H). |
|
|
Description for D33 4-Hydroxy-2-methylbenzonitrile (D33)To a solution of 2-methyl-4-(methyloxy)benzo?iotat?le (7 g) in anhydrous DCM (100 mL) was added drapwise BBr3 (51 5 g) at -78 C The resulting mixture was allowed to warm to RT and stirred for 24 hours LCMS indicated the reaction was completed Water was added dropwise slowly to quech the reaction. The mixture was extracted with EA (3x100 mL) and the combined organic layer was washed with brine dried over sodium sulfate, and concentrated to give 5 4 g of 4-hydroxy-2-methylbenzo nitrile (D33) as a white solid. deltaH (DMSO-Cf6, 400MHz). 2.45(3H, s), 6.67(1H1 d), 6 69(1H1 d), 7 51(1H, d), 1043(1H1 s). MS (ES) C8H7NO requires 133, found 134 1 (M+ H+) |
|
With decylthiol; potassium tert-butylate; In N,N-dimethyl-formamide; at 110℃; for 3h; |
Decanethiol (261 mg, 1.5 mmol) and t-BuOK (168 mg, 1.5 mmol) were added to a solution of <strong>[21883-13-6]4-methoxy-2-methylbenzonitrile</strong> (147 mg, 1 mmol) in DMF (5 mL). The reaction mixture was stirred at 110 C for 3 h. The mixture was then diluted with water (30 mL) and extracted with EtOAc (10 mL x 3). The extracts were washed with brine (10 mL x 3), dried over Na2SO4, and concentrated under reduced pressure. The resulting residue was purified by silica gel column with (petroleum ether:EtOAc = 10:1) to provide 4-hydroxy-2-methylbenzonitrile (70 mg, yield: 52.6%). LC-MS (011): 134.70 [M+H]+; Rt: 1.44 min, Purity: 80% (254 nm). |
|
|
A solution of <strong>[21883-13-6]4-methoxy-2-methylbenzonitrile</strong> (5.O g, 33.97 mmol) in DCM (15O mL) is cooled down to 00C before adding dropwise a 1IW solution of BBr3 in DCM (136 mL, 136 mmol). The reaction mixture is allowed to reach rt and stirring is then continued at 45C for 5 days. Ice water (500 mL) is then added and the reaction mixture is stirred for 1 h before sat. aq. NaHCO3 (250 mL) is added. The mixture is extracted wit DCM (200 mL then 4 x 100 mL) and the combined org. extracts are dried over MgSO4, filtered and evaporated to give the title compound as a brown solid (4.7 g). LC-MS: tR = 0.76 min. 1H NMR (D6-DMSO): delta 2.38 (s, 3H), 6.73 (dd, J = 8.5, 2.0 Hz, 1 H), 6.79 (d, J = 2.0 Hz, 1 H), 7.55 (d, J = 8.5 Hz, 1 H), 10.49 (s, 1 H). |






 Chemistry
Chemistry
 Pharmaceutical Intermediates
Pharmaceutical Intermediates
 Inhibitors/Agonists
Inhibitors/Agonists
 Material Science
Material Science







 For Research Only
For Research Only
 120K+ Compounds
120K+ Compounds
 Competitive Price
Competitive Price
 1-2 Day Shipping
1-2 Day Shipping