| 86% |
|
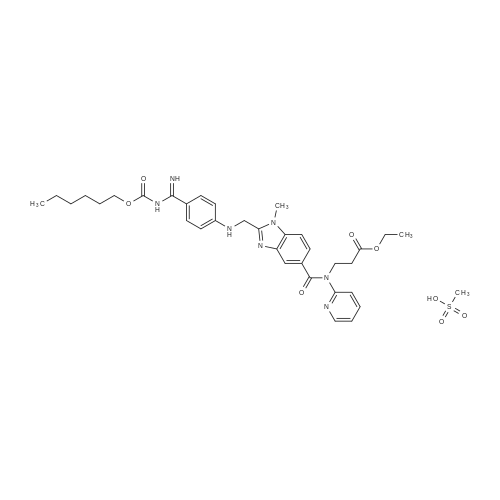
This embodiment's darbey adds the group ester method for the preparation of intermediates with the following steps:(1) under the protection of nitrogen, the 28.3g the N-(4-cyano-phenyl)-glycine (0.16mol) and 25.9g condensing agent CDI (0.16mol) dissolved in the 100 ml of methylene chloride and 100 ml of a mixed solvent of the ether in a, stirring and heating to 40 ±2 C reflux, thermal insulation reaction 1-2h.(2) added to the above-mentioned reaction system 50.0g of 3-amino-4-methyl amino-benzoic acid-N-(2-pyridyl)-N - (2-ethoxy carbonyl-ethyl)-amide (0.14mol), in 40 ±2 C continue to return to the temperature of the condensation reaction, to 3-amino-4-methyl amino-benzoic acid-N-(2-pyridyl)-N - (2-ethoxy carbonyl-ethyl)-amide reaction is complete, to stop the reaction.Generating compound IV does not need separation directly used for the next step.(3) in the above-mentioned reaction system is added to 160 ml of acetic acid, first decompression concentrating evaporating methylene chloride and ethyl ether, then heating to 50-60 C to cyclization reaction, until the compound IV the reaction is complete, to stop the reaction.(4) after the reaction, water is poured in the reaction system, cooling to 20-30C, crystallization, filtration, the filter cake after re-crystallization with ethanol, again filtered, the filter cake is drying to obtain 60.6g compound I, to yield 86.0%, |
| 85% |
|
(1) at room temperature,75 g of compound of formula 6,1000 mL THF,100mL triethylamine mixed,Cooled to 0 ,70mL pivaloyl chloride was added below 10 and the reaction was incubated for 0.5h; After the reaction was completed, 155g of compound of formula 4 was added,45 reaction 2h;The organic layer was evaporated to dryness and added to 1500mL n-butyl acetate, at 90 for 4h,700 mL of butyl acetate was distilled off under reduced pressure,The crystals were stirred at 5 C,Filtered and dried to give a dry product of formula 3 175g,The yield is 85%; |
| 83% |
|
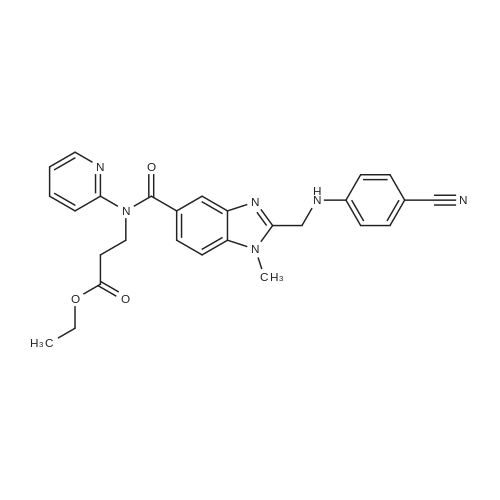
4-Nitriloanilino-acetic acid (2.32 g, 13.2 mmol) was dissolved in 80 ml of DMF,1-Hydroxybenzotriazole (H0BT) (1.96 g, 14.5 mmol) was added,Ethyl-3- (3-dimethylaminopropyl) carbodiimide hydrochloride (EDCI) (2.77 g, 14.5 mmol) was added at 0 C under stirring for 45 min,Slowly rise to room temperature,Ethyl 3- (1-methylamino-2-amino-phenyl-4-yl) -carboxylic acid- (N-2-pyridyl) amidopropionic acid ethyl ester (5.50111111.01) was added at room temperature the reaction overnight, concentrated, diluted with ethyl acetate and large, washed three times with brine, dried over Na2S04 after dried, concentrated and the crude product in 60ml of acetic acid was refluxed for 1.5h, was concentrated, basified added 1.5N aqueous ammonia, extracted three times with ethyl acetate. (60 mL X 3). The organic phase was washed once with brine, dried over Na 2 SO 4 and concentrated. The crude product was purified by silica gel column chromatography.The amorphous solid, 3- (1-methyl-2- (4-nitrile-phenylaminomethyl) -5-yl-carboxylic acid- (N-2-pyridyl) amido) propionic acid ethyl ester (6.4 g, yield 83%). Mass spectrum (ESI-MS): 482 ? 1 (M + H) +, 505.4 (M + Na) +; C27H26N6O3 (482) |
| 61% |
|
a) 1-Methyl-2-[N-(4-cyanophenyl)-aminomethyl]-benzimidazol-5-yl-carboxylic acid-N-(2-pyridyl)-N-(2-ethoxycarbonylethyl)-amide Prepared analogously to Example 25c from N-(4-cyanophenyl)-glycine and 3-amino-4-methylamino-benzoic acid-N-(2-pyridyl)-N-(2-ethoxycarbonylethyl)-amide. Yield: 61% of theory, Rf value: 0.62 (silica gel; dichloromethane/methanol=19:1) |
|
|
Example 1 - Large-scale industrial synthesis of the compound of formula 4-Br88 kg carbonyl-di-(l,2,4-triazole) are taken and combined with 920 1 tetrahydrofuran. The contents of the apparatus are heated to 35C with stirring. Then 90 kg of compound 3 are added batchwise at 35C within 1 to 2 hours.160 kg of compound 2 are placed in a second reaction vessel, then 350 1 tetrahydrofuran are added and the mixture is heated to 500C with stirring.The solution of 3 is metered into the solution of 2 within 2 to 3 hours at 47C - 53C and the solution obtained is diluted with 115 1 tetrahydrofuran. Then the mixture is stirred for another 4 hours at 47C - 53C ( preferably 500C ). Then 670 1- 695 1 tetrahydrofuran are distilled off in vacuo at 50C-60C. 235 1 of n-butyl acetate are then allowed to flow into the residue. After this, 600 1 -630 1 of a butyl acetate/THF mixture are distilled off in vacuo at 50C-85C. During the distillation 700 1 butyl acetate are metered in. 65 kg acetic acid are allowed to flow into the residue, the contents are heated to 85C-90C and stirred for at least another 2.5 h at this temperature. Then the mixture is cooled to 65C-75C. A solution of 165 1 water and 20 kg common salt is added to the contents and the mixture is rinsed with 300 1 water. Then the temperature is adjusted to 60C-70C and the mixture is stirred for a minimum of 15 min. at this temperature. For phase separation the stirrer is stopped and the mixture is left to settle for at least 15 min. The aqueous phase is drained off into another reaction vessel which contains 120 1 of n-butyl acetate. The mixture is heated to 60C-70C with stirring and stirred for at least 10 min. After phase separation the aqueous phase is drained off into the chemical waste drain. The butyl acetate phases and 20 1 of butyl acetate for rinsing are combined. 590 1 - 620 1 of n-butyl acetate are distilled off from this content in vacuo at a max. internal temperature of 800C. <n="12"/>880 1 isopropanol are allowed to flow into the distillation residue and the content is adjusted to 32C-38C. Then approx. 90 kg of 48% hydrobromic acid are metered in at 32C-38C until the pH value is 0.6 to 1.3. The mixture is stirred for a minimum of 20 min. at 32C-38C and then cooled to 7C-13C and stirred at this temperature for at least one hour. The resulting suspension is centrifuged, washed with a total of 840 1 isopropanol and dried in vacuo at max. 55C. Yield: 211 kg-250 kg. Mp.: 200-2150C (with decomposition).The compound 4-HBr may be isolated using any standard commercially available centrifuge. |
|
|
Example 1 : Ethyl N-[(2-[(p-cyanophenyl)amino1methyl)-1 -methyl-1 H- benzimidazole-5-carbonyl1-N-(2-pyridyl)-3-aminopropionate hydrochloride (IV-HCI)Under Ar atmosphere, A/-(p-cyanophenyl)-glycine (V) (7.22 g, 41 .0 mmol) and 1 ,1 '-carbonyldiimidazole (6.64 g, 41 .0 mmol) were suspended in anhydrous THF (315 mL). It was refluxed for 45 minutes and a solution of compound (VI) (12.74 g, 37.2 mmol) in anhydrous THF (56 mL) was added slowly. After 6 h under reflux, the reaction mixture was cooled down, and the solvent was distilled under low pressure. The oil obtained was dissolved in glacial acetic acid (155 mL) and refluxed for 1 h. The solvent was removed under low pressure, the residue was dissolved in CH2CI2 (130 mL) and washed with H2O (2 x 130 mL). The organic phase was dried over anhydrous MgSO4 and the solvent was distilled under low pressure. The residue obtained was dried under vacuum, obtaining 20.19 g of crude (IV) (75% a/a purity according to HPLC/MS).The brown oil was dissolved in isopropanol (101 mL) at 35 C and HCI(c) (37%, 3.40 mL, 41 .1 mmol) was added slowly. After a short time an abundant white solid appeared. It was stirred at 35 C for 30 min and next at 0 C for 30 min. The solid was filtered out, washed with IPA (15 mL) and dried under vacuum, obtaining the product (IV-HCI) (14.25 g, 74% yield, 97% a/a purity according to HPLC/MS). This solid was recrystallized from EtOH (160 mL), washed with EtOH (2 x 10 mL) and dried under vacuum, obtaining 12.13 g (23.4 mmol, 63% global yield, 99% a/a purity according to HPLC/MS) of the product (IV-HCI).1H RMN (400 MHz, CD3OD) : delta (ppm) = 8.33 (ddd, J = 4.8, 2.0, 0.8, 1 H), 7.77 (d, J = 8.8, 1 H), 7.66 (dd, J = 1 .6, 0.8, 1 H), 7.60 (ddd, J = 8.0, 8.0, 2.0, 1 H), 7.55-7.50 (m, 3H), 7.17 (ddd, J = 7.6, 4.8, 0.8, 1 H), 7.09 (d, J = 8.0, 1 H), 6.83 (d, J = 8.8, 2H), 5.02 (s, 2H), 4.34 (t, J = 7.2, 2H), 4.05 (q, J = 7.2, 2H), 4.02 (s, 3H), 2.76 (t, J = 7.2, 2H), 1 .19 (t, J = 7.2, 3H).Melting point (Tme,t): 213-215 CPXRD: FIG. 2, 2theta angle values () = 3.7, 10.0, 10.9, 17.7, 18.3, 20.9, 22.0, 22.5, 23.7, 25.9, 26.4. |
|
|
Example-20: Preparation of l-methyI-2-[N-(4-cyanophenyl) aminomethyl] benzimidazol-5-ylcarboxylicacid-N-(2-pyridyl)-N-(2-ethoxycarbonylethyl)amide compound of formula-14 A mixture of 2-(4-cyanophenylamino)acetic acid (139 g) and tetrahydrofuran (750 ml) was heated to 50-55C. A solution of N,N'-carbonyldiimidazole (177.6 g) in tetrahydrofuran (1000 ml) was added to the above reaction mixture at the same temperature over a period of 1 hour and stirred for 2 hours at 50-55C. A solution of ethyl 3-(3-amino-4-(methylamino)-N-(pyridin-2-yl)benzamido) propanoate compound of formula- 12 (250 g) in tetrahydrofuran (1500 ml) was added over a period of 2 hours at 50-55C and heated to 60-65C. The reaction mixture was stirred for 50 hours at 60- 65C. After completion of the reaction, distilled off the solvent from the reaction mixture. Acetic acid was added to the reaction mixture and heated to 95-100C. The reaction mixture was stirred for 5 hours at 95-100C. Distilled off the solvent completely under the reduced pressure and the reaction mixture was cooled to 25-30C. Water was added to the reaction mixture and the product was extracted with dichloromethane. Dichloromethane layer was washed with water followed by sodium chloride. Distilled off the solvent completely from the dichloromethane layer to obtain title compound.Yield: 300 g |
|
|
Example-4: Process for preparation of the crystalline ethyl 3-(2-((4-cvanophenylamino)methylVl- methvl-N-(pvridin-2-vO-lH-benzotdUmidazole-5-carboxamido) propanoate of Formula (2B) 77 g 2-(4-cyanophenylamino) acetic acid of Formula (E), 71.04 g CDI in 1000 mL Toluene were stirred at 25C to 35C. The reaction mixture was heated to 55C to 60C and maintained for 2 hours. 100 g ethyl 3-(3-amino-4-(methylamino)-N-(pyridin-2- yl)benzamido)propanoate of Formula (2A) was added to the reaction mixture and stirred for 3 hours at 60C to 65C. The reaction mixture was further heated at 100C for 3 hours. Toluene was distilled under vacuum. The residue was treated with 500 mL methylene dichloride and organic layer was washed with water. The organic layer was filtered and washed with methylene dichloride. The methylene dichloride was distilled under vacuum and 100 mL ethyl acetate was added at 40C to 45C and stirred for 15 minutes. The reaction mixture was heated to 75C and cooled to 10C to 15C. The reaction mixture was stirred for 2 hours and the precipitated product was filtered and washed with 150 mL ethyl acetate to obtain crystalline ethyl 3-(2-((4-cyanophenylamino)methyl)-l-methyl-N- (pyridin-2-yl)-lH-benzo[d]imidazole-5-carboxamido) propanoate of Formula (2B). The compound of Formula (2B) is characterized by x-ray powder diffraction as depicted in Fig.2. |
| 300 g |
|
A mixture of 2-(4-cyanophenylamino)acetic acid (139 g) and tetrahydrofuran (750 ml) was heated to 50-55 C. A solution of N,N?-carbonyldiimidazole (177.6 g) in tetrahydrofuran (1000 ml) was added to the above reaction mixture at the same temperature over a period of 1 hour and stirred for 2 hours at 50-55 C. A solution of ethyl 3-(3-amino-4-(methylamino)-N-(pyridin-2-yl)benzamido) propanoate compound of formula-12 (250 g) in tetrahydrofuran (1500 ml) was added over a period of 2 hours at 50-55 C. and heated to 60-65 C. The reaction mixture was stirred for 50 hours at 60-65 C. After completion of the reaction, distilled off the solvent from the reaction mixture. Acetic acid was added to the reaction mixture and heated to 95-100 C. The reaction mixture was stirred for 5 hours at 95-100 C. Distilled off the solvent completely under the reduced pressure and the reaction mixture was cooled to 25-30 C. Water was added to the reaction mixture and the product was extracted with dichloromethane. Dichloromethane layer was washed with water followed by sodium chloride. Distilled off the solvent completely from the dichloromethane layer to obtain title compound. |
| 127 g |
|
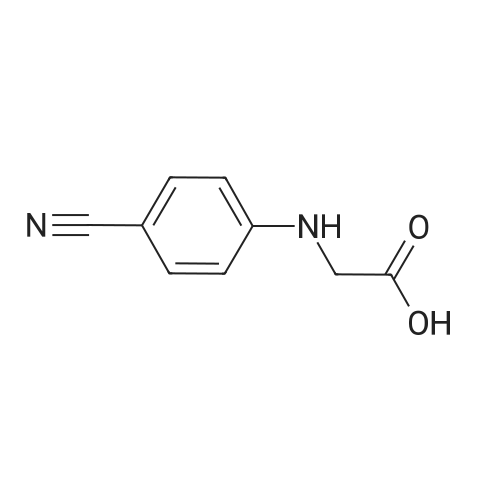
A mixture of N-(4-cyanophenyl) glycine (56.6 g), CDI (61.6 g) and imidazole*HC1 (7.6g) in anhydrous EtOAc (1000 mL), was stirred for lh at 45-50C, under inert atmosphere. phenylene-l,2-diamine of formula 2 (lOOg) was, lot-wise, charged to the reaction mass. After stirring for another lh at the same temp, Glacial acetic acid (100mL) was added and the temperature of the reaction mass maintained at reflux for another6h. The reaction mixture cooled to 45-50C, and washed, successively with water and brine solution. The separated organic layer was concentrated under vacuum, till approx. 3 volumes were left behind. The reaction mass was cooled to room temp and 5% Brine solution (500 mL) was charged and the precipitate obtained was slurred for 3h. Theprecipitate was filtered, washed with EtOAc and dried under vacuum at 50C, to afford compound of formula 3 as off-white solid material (127.Og, 98.8% HPL pure). |
|
|
77 g 2-(4-cyanophenylamino) acetic acid of Formula (E), 71.04 g CDI in 1000 mL Toluene were stirred at 25 C. to 35 C. The reaction mixture was heated to 55 C. to 60 C. and maintained for 2 hours. 100 g ethyl 3-(3-amino-4-(methylamino)-N-(pyridin-2-yl)benzamido)propanoate of Formula (2A) was added to the reaction mixture and stirred for 3 hours at 60 C. to 65 C. The reaction mixture was further heated at 100 C. for 3 hours. Toluene was distilled under vacuum. The residue was treated with 500 mL methylene dichloride and organic layer was washed with water. The organic layer was filtered and washed with methylene dichloride. The methylene dichloride was distilled under vacuum and 100 mL ethyl acetate was added at 40 C. to 45 C. and stirred for 15 minutes. The reaction mixture was heated to 75 C. and cooled to 10 C. to 15 C. The reaction mixture was stirred for 2 hours and the precipitated product was filtered and washed with 150 mL ethyl acetate to obtain crystalline ethyl 3-(2-((4-cyanophenylamino)methyl)-1-methyl-N-(pyridin-2-yl)-1H-benzo[d]imidazole-5-carboxamido) propanoate of Formula (2B). The compound of Formula (2B) is characterized by x-ray powder diffraction as depicted in FIG.2. |
|
|
1 kg of organic solvent a, 1.5 kg of SM2, and 1.2 kg of CDI were sequentially added to the reaction vessel, and the reaction was stirred at 10 to 50 C for 3 hours.Then, 1 kg of SM1 and 1 kg of organic solvent b were sequentially added, and the reaction was further stirred for 12 hours, and the solvent was distilled off under reduced pressure at 40 C to the reaction kettle.The acetic acid was put into the mixture, heated to 110 C for 5 hours, and the solvent was distilled off under reduced pressure at 100 C or less.Washing with water, recovering the solvent under reduced pressure in the organic phase, adding ethyl acetate to the residue, stirring to dissolve, and adding oxalic acidThe hydrate solution was stirred and crystallized at 22 C for 2 hours, centrifuged, and the filter cake was washed with ethyl acetate. The filter cake was dried under reduced pressure at 45 C for 6 hours.When, a pale yellow solid PR-I;Wherein, the solution of oxalic acid dihydrate is an ethanol solution containing oxalic acid dihydrate, and the oxalic acid dihydrate is in solution.The part by mass is 10%.Wherein, the organic solvent a and the organic solvent b are both N,N-dimethylformamide;Among them, the mass ratio of other materials is SM1: acetic acid = 1:8, SM1: dichloromethane = 1:3, dichloromethane: water =1:3, SM1: solution containing oxalic acid dihydrate = 1:5; |






 Chemistry
Chemistry
 Pharmaceutical Intermediates
Pharmaceutical Intermediates
 Inhibitors/Agonists
Inhibitors/Agonists
 Material Science
Material Science







 For Research Only
For Research Only
 120K+ Compounds
120K+ Compounds
 Competitive Price
Competitive Price
 1-2 Day Shipping
1-2 Day Shipping