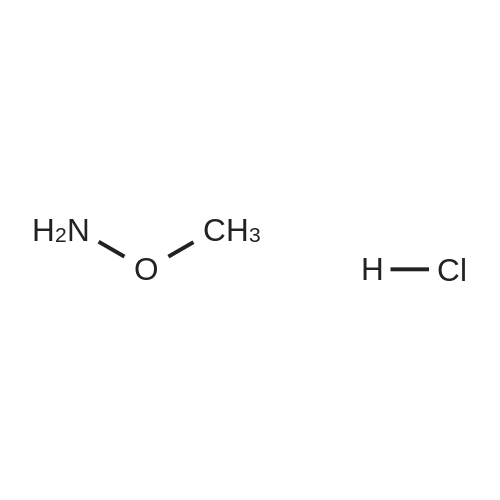
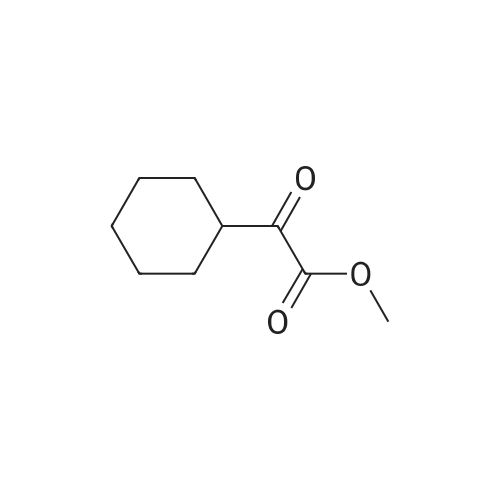
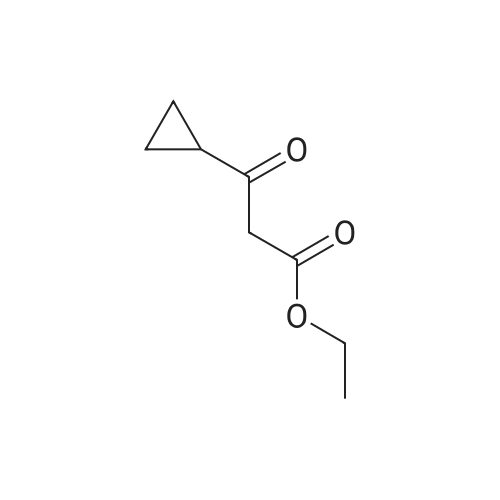
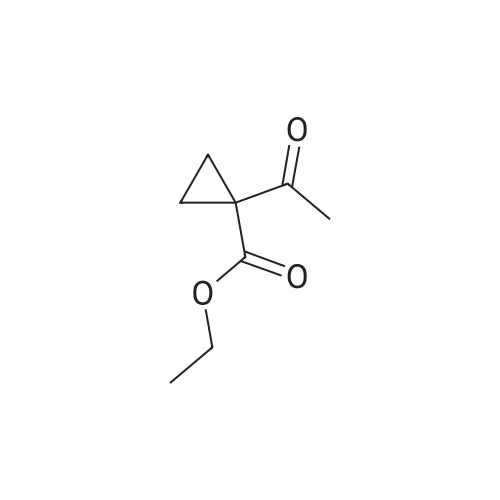
| 74% |
With sodium; In ethanol; for 2.5h;Reflux; |
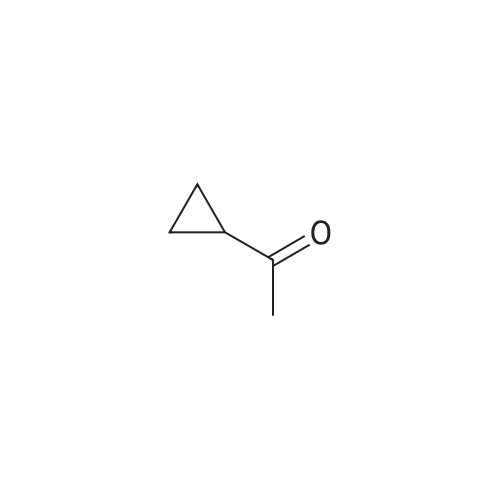
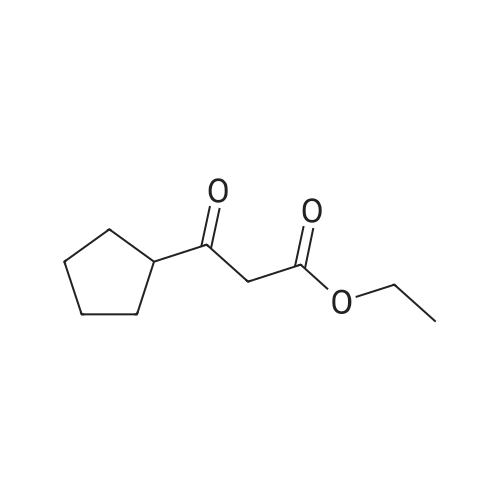
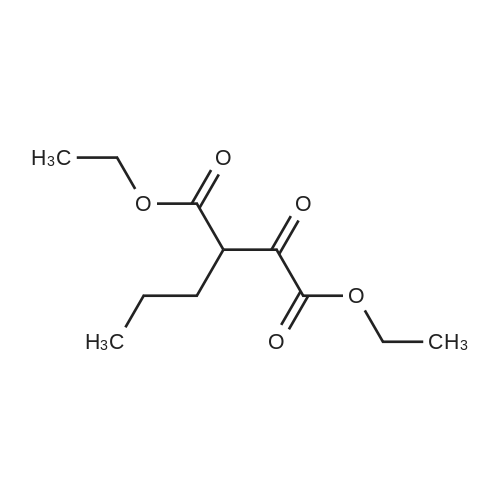
Intermediate 29ethyl 4-cyclopropyl-2,4-dioxobutanoateSodium metal (2.411 g, 105 mmol) was dissolved in ethanol (50 mL). The solution was heated to reflux followed by addition of a mixture of 1-cyclopropylethanone (8.4 g, 100 mmol) and diethyl oxalate (14.59 g, 100 mmol) dropwise over 30 minutes. The reaction mixture was heated at reflux for an additional 2h, and then allowed to cool to room temperature over a 2 d period. The contents were diluted with water (200 mL) and acidified by dropwise addition of 6N HC1. The contents were extracted with EtOAc (3 x 75 mL), washed with water, brine, dried over MgS04, filtered, and concentrated in vacuo. The final product was collected as 14.3g (74%). LCMS E-S (M+H) =184.8 1H NMR (400 MHz, CHLOROFORM-d) delta ppm 1.03 (d, J=7.83 Hz, 2 H) 1.13 - 1.19 (m, 2 H) 1.31 (t, J=7.07 Hz, 3 H) 1.81 - 1.90 (m, 1 H) 4.29 (q, J=7.16 Hz, 2 H) 6.43 (s, 1 H). |
| 66% |
With sodium ethanolate; In ethanol; at 0 - 80℃; |
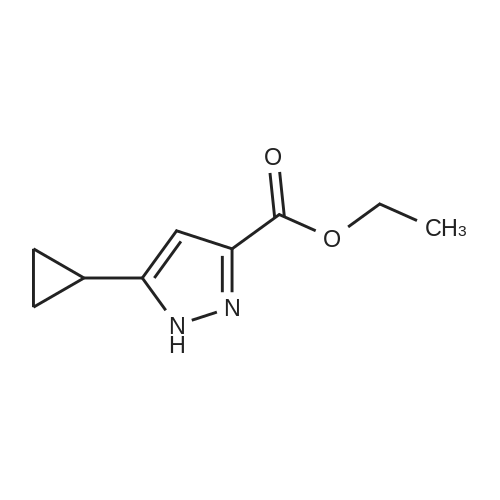
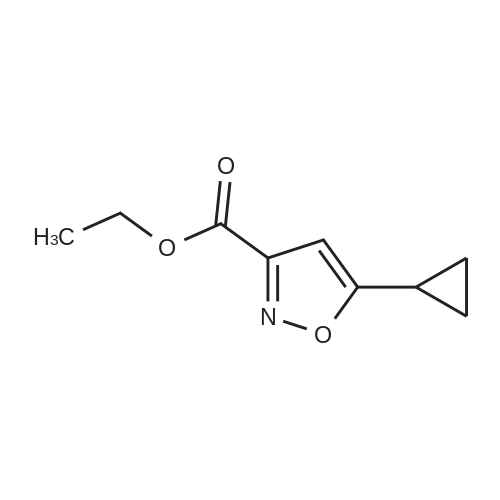
Sodium ethoxide (10 g, 147 mmol) is combined with absolute EtOH (60 mL) in an oven-dried flask, under nitrogen and heated to 70 C. to aid dissolution. The mixture is cooled to 0 C., treated drop-wise with a mixture of cyclopropyl methyl ketone (14.56 mL, 147 mmol) and diethyl oxylate (19.96 mL, 147 mmol) and warmed to RT. Stirring was difficult, so additional EtOH (60 mL) is added and the mixture is stirred for 1 h, then heated to 80 C. for 45 min. The mixture is cooled to RT and concentrated to dryness. The resulting solid is triturated with EtOAc, filtered, and rinsed with EtOAc and Et2O to remove the reddish color. The solid is dissolved in water (300 mL), acidified to pH 2 with dilute H2SO4, extracted with Et2O (400 mL total), dried (Na2SO4) and concentrated to afford 18.0 g (66% yield) of ethyl 4-cyclopropyl-2,4-dioxobutanoate as an amber oil. HRMS (ESI) calcd for C9H12O4 +H: 185.0814, found 185.0821 (M+H)+. Ethyl 4-cyclopropyl-2,4-dioxobutanoate (12.92 g, 70.1 mmol) is combined with hydroxylamine hydrochloride (14.62 g, 210.4 mmol) in EtOH (250 mL), heated to reflux for 1 h, cooled, and concentrated to dryness. The residue is partitioned between H2O (250 mL) and EtOAc (2×250 mL) and the combined organics are dried (MgSO4) and concentrated to an amber oil (13.89 g). The crude material is chromatographed over 500 g silica gel, eluting with 25% EtOAc/hexane. The appropriate fractions are combined and concentrated to afford 10.71 g (84% yield) of ethyl 5-cyclopropylisoxazole-3-carboxylate as a yellow oil. MS (CI) m/z: 182 (M+H)+. Sodium hydroxide (1.76 g, 44.0 mmol) in H2O (5 mL) is added to a solution of ethyl 5-cyclopropylisoxazole-3-carboxylate (1.97 g, 10.9 mmol) in MeOH (10 mL). The mixture is stirred at RT for 3 h, concentrated to remove the MeOH, and acidified to pH 2 with 5% HCl. The acid is extracted with CH2Cl2 (6×20 mL), dried (MgSO4) and concentrated to afford 1.56 g (93% yield) of 5-cyclopropylisoxazole-3-carboxylic acid as a white solid. MS (Cl) m/z: 154 (M+H)+. 5-Cyclopropylisoxazole-3-carboxylic acid (1.53 g, 10 mmol) is dissolved in benzene (30 mL), treated with oxalyl chloride (3.46 mL, 40 mmol) and heated to reflux for 2 h. The mixture is cooled, concentrated to dryness and the residual benzene is azeotroped off with CH2Cl2. The resulting acid chloride is dissolved in Me2CO (15 mL) and treated with a solution of NaN3 (1.95 g, 30 mmol) in H2O (7 mL). The mixture is vigorously stirred for 1 h, concentrated to remove the Me2CO, triturated with H2O, filtered, rinsed with water and dried under vacuum to afford 1.76 g (99% yield) of 5-cyclopropylisoxazole-3-carbonyl azide as an off-white solid. 1H NMR (CDCl3, 400 MHz): delta 1.02, 1.14, 2.10, 6.35 ppm. 5-Cyclopropylisoxazole-3-carbonyl azide (447 mg, 2.5 mmol) is combined with 5-chloro-2,4-dimethoxyaniline (471 mg, 2.5 mmol) in anhydrous MeCN (30 mL) and heated to reflux for 18 h. The mixture is cooled and the resulting solid is filtered, rinsed with Et2O and dried in a vacuum oven to afford 619 mg (73% yield) of Example 621 as a very light purple solid. HRMS (ESI) calcd for C15H16N3O4Cl +H: 338.0907, found 338.0896 (M+H)+. |
| 48% |
|
Metal Na (6 g, 0.26 mol) was added to ethanol (1 .5 L) in portions and stirred until it dissolved. The above solution was cooled in an ice-water bath, and compound 5 (20 g, 0.24 mol) was added dropwise under N2 atmosphere. 0.5 h later, oxalic acid diethyl ester (31 .6 g, 0.22 mol) was added dropwise at 0C with strong stirring. The resulting reaction mixture was stirred at room temperature overnight. The mixture was diluted with water and concentrated to remove ethanol on a rotary evaporator (water bath be- low 40 C), and the residue was acidified to pH 2 by 6N aq. HCI and extracted with DCM. The combined extracts were washed with brine, dried over anhydrous Na2S04 and concentrated on a rotary evaporator (water bath below 30 C) to give compound 6 (21 g, yield: 48%). H-NMR (CDCI3): delta= 6.47 (s, 1 H), 4.37-4.32 (m, 2H), 1.92-1.86 (m, 1 H), 1 .37 (t, 3H, J= 7.2 Hz), 1 .24-1 .04 (m, 4H). |
| 31% |
With sodium ethanolate; In ethanol; at 0 - 20℃; |
To a solution of sodium ethoxide (freshly prepared by dissolving sodium (1 g, 8.2 mmol, 1.2 eq) in EtOH (30 mL)), diethyl oxalate (0.92 mL, 6.85 mmol, 1 eq) was added at room temperature followed by addition of cyclopropyl methyl ketone (0.74 mL, 7.5 mmol, 1.1 eq) dropwise at 00C. The reaction mixture was slowly warmed to room temperature and stirred for 3 h. Ice cold water (10 mL) was added and EtOH was evaporated under reduced pressure. The residual aqueous layer was diluted with 2 N aq. HCI (15mL) and extracted with diethyl ether (2 * 25 mL). The organic layer was washed with brine solution and dried (Na2SO4), filtered and concentrated to give a pale brown liquid (400 mg, 31%). |
| 31% |
With sodium methylate; In ethanol; at 0 - 20℃; for 3h; |
Diethyl oxalate (0.92 mL, 6.85 mmol, 1 equiv.) was added at RT to a freshly prepared sodium methanolate solution (prepared by dissolving sodium (1 g, 8.2 mmol, 1.2 equiv.) in EtOH (30 mL)), and cyclopropyl methyl ketone (0.74 mL, 7.5 mmol, 1.1 equiv.) was then added dropwise at 0 C. The reaction mixture was slowly heated to RT and stirred for a further 3 h. Ice-cold water (10 mL) was added and the EtOH was distilled off under reduced pressure. The remaining aqueous phase was diluted with 2N hydrochloric acid (15 mL) and extracted with diethyl ether (2×25 mL). The combined organic phases were washed with saturated sodium chloride solution, dried over Na2SO and concentrated under reduced pressure. A pale brown liquid was obtained as product (400 mg, 31% yield). |
| 31% |
With ethanol; sodium; at 0 - 23℃; for 3h; |
To a solution of sodium ethoxide (freshly prepared by dissolving sodium (1 g, 8.2 mmol, 1.2 equivalents) in ethanol (30 mL)), diethyl oxalate (0.92 mL, 6.85 mmol, 1 equivalent) was added at room temperature followed by addition of cyclopropyl methyl ketone (0.74 mL, 7.5 mmol, 1.1 equivalents) dropwise at 0 C. The reaction mixture was slowly warmed to room temperature and stirred for 3 h. Ice cold water (10 mL) was added and ethanol was evaporated under reduced pressure. The residual aqueous layer was diluted with 2 N aq. HCI (15mL) and extracted with diethyl ether (2 * 25 mL). The organic layer was washed with brine solution and dried over sodium sulphate, filtered and concentrated to give a pale brown liquid (400 mg, 31 %). |
| 31% |
With sodium ethanolate; at 0 - 20℃; for 3h; |
Step a: To a solution of sodium ethoxide (freshly prepared by dissolving sodium (1 g, 8.2 mmol, 1.2 equivalents) in ethanol (30 mL)), diethyl oxalate (0.92 mL, 6.85 mmol, 1 equivalent) was added at room temperature followed by addition of cyclopropyl methyl ketone (0.74 mL, 7.5 mmol, 1.1 equivalents) dropwise at 0? C. The reaction mixture was slowly warmed to room temperature and stirred for 3 h. Ice cold water (10 mL) was added and ethanol was evaporated under reduced pressure. The residual aqueous layer was diluted with 2 N aq. HCl (15 mL) and extracted with diethyl ether (2?-25 mL). The organic layer was washed with brine solution and dried over sodium sulfate, filtered and concentrated to give a pale brown liquid (400 mg, 31%). |
| 31% |
With sodium ethanolate; In ethanol; at 0 - 20℃; for 3h; |
To a solution of sodium ethoxide (freshly prepared by dissolving sodium (1 g, 8.2 mmol, 1 .2 equivalents) in ethanol (30 mL)), diethyl oxalate (0.92 mL, 6.85 mmol, 1 equivalent) was added at room temperature followed by addition of cyclopropyl methyl ketone (0.74 mL, 7.5 mmol, 1 .1 equivalents) dropwise at 0 C. The reaction mixture was slowly warmed to room temperature and stirred for 3 h. Ice cold water (10 mL) was added and ethanol was evaporated under reduced pressure. The residual aqueous layer was diluted with 2 N aq. HCI (15mL) and extracted with diethyl ether (2 ? 25 mL). The organic layer was washed with brine solution and dried over sodium sulphate, filtered and concentrated to give a pale brown liquid (400 mg, 31 %). |
| 31% |
With sodium ethanolate; at 0 - 23℃; for 3h; |
Step a: To a solution of sodium ethoxide (freshly prepared by dissolving sodium (1 g, 8.2 mmol, 1 .2 equivalents) in ethanol (30 mL)), diethyl oxalate (0.92 mL, 6.85 mmol, 1 equivalent) was added at room temperature followed by addition of cyclopropyl methyl ketone (0.74 mL, 7.5 mmol, 1 .1 equivalents) dropwise at 0 C. The reaction mixture was slowly warmed to room temperature and stirred for 3 h. Ice cold water (10 mL) was added and ethanol was evaporated under reduced pressure. The residual aqueous layer was diluted with 2 N aq. HCI (15mL) and extracted with diethyl ether (2 chi 25 mL). The organic layer was washed with brine solution and dried over sodium sulphate, filtered and concentrated to give a pale brown liquid (400 mg, 31 %). |
| 31% |
With sodium ethanolate; In ethanol; at 0 - 23℃; for 3h; |
To a solution of sodium ethoxide (freshly prepared by dissolving sodium (1 g, 8.2 mmol, 1 .2 equivalents) in ethanol (30 mL)), diethyl oxalate (0.92 mL, 6.85 mmol, 1 equivalent) was added at room temperature followed by addition of cyclopropyl methyl ketone (0.74 mL, 7.5 mmol, 1 .1 equivalents) dropwise at 0 '?. The reaction mixture was slowly warmed to room temperature and stirred for 3 h. Ice cold water (10 mL) was added and ethanol was evaporated under reduced pressure. The residual aqueous layer was diluted with 2 N aq. HCI (15mL) and extracted with diethyl ether (2 chi 25 mL). The organic layer was washed with brine solution and dried over sodium sulphate, filtered and concentrated to give a pale brown liquid (400 mg, 31 %). |
| 31% |
With sodium ethanolate; at 0 - 23℃; for 3h; |
Step a: To a solution of sodium ethoxide (freshly prepared by dissolving sodium (1 g, 8.2 mmol, 1 .2 equivalents) in ethanol (30 mL)), diethyl oxalate (0.92 mL, 6.85 mmol, 1 equivalent) was added at room temperature followed by addition of cyclopropyl methyl ketone (0.74 mL, 7.5 mmol, 1 .1 equivalents) dropwise at 0 C. The reaction mixture was slowly warmed to room temperature and stirred for 3 h. Ice cold water (10 mL) was added and ethanol was evaporated under reduced pressure. The residual aqueous layer was diluted with 2 N aq. HCL (15mL) and extracted with diethyl ether (2 chi 25 mL). The organic layer was washed with brine solution and dried over sodium sulphate, filtered and concentrated to give a pale brown liquid (400 mg, 31 %) |
|
With potassium tert-butylate; In tetrahydrofuran; at 20℃; |
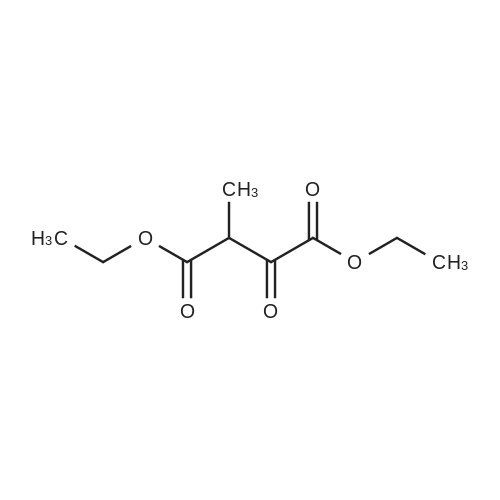
General procedure: To a mixture of t-BuOK (92.0 g, 697 mmol) and THF (500 mL) was added a mixture of 3-methyl-2-butanone (50.0 g, 581 mmol) and diethyl oxalate (85.8 g, 587 mmol) dropwise over 1 h. The mixture was stirred at room temperature for 15 h to form ethyl 5-methyl-2,4-dioxohexanoate. Thereafter, AcOH (80 mL, 1.39 mol) and hydrazine hydrate (32.0 g, 639 mmol) were successively added. The mixture was heated under reflux with stirring for 2 h. After being cooled, the mixture was concentrated in vacuo and the residue was partitioned between EtOAc and water. The organic solution was washed with sat. NaHCO3 and brine, dried over MgSO4, and concentrated under reduced pressure. The residue was purified by silica gel chromatography (hexane/EtOAc, 9:1 to 1:1) to give 20a (57.7 g, 55%) as a brown oil. |
|
With sodium ethanolate; In ethanol; at 0 - 80℃; |
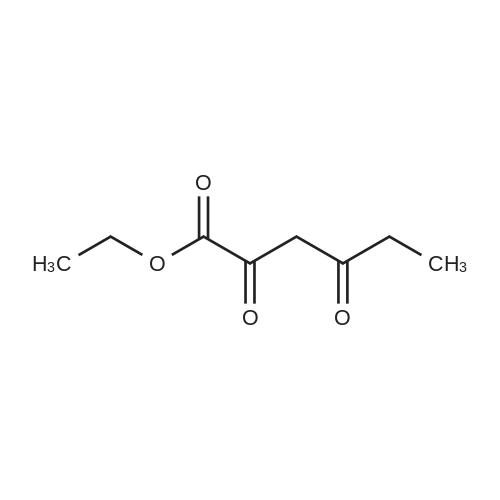
Sodium metal was dissolved into a solution of EtOH (150ml) at RT under nitrogen atmosphere to form NaOEt (16.19 gm). This mixture was cooled to 0 C. Diethyl oxalate (34.76gm) and isopropyl methyl ketone (20gm) was added drop wise for about 15 min and warmed to RT. Now EtOH (100 ml) was added and stirred at RT for about 1 hour. Heat this reaction mixture to 80 C for about 45 minuets and cooled to RT and concentrated under reduced pressure. To this resulting solid, add EtOAC. Wash with EtOH and filtered on cloth to get fine smooth powder.. This solid is dissolved in water and acidified with dilute Sulphuric acid (pH-2). This compound is extracted with diethyl ether and dried over sodium sulphate and was concentrated under reduced pressure to obtain the brown colored liquid compound (40g, 93% yield ). |
|
With ethanol; sodium; at 0 - 80℃; for 2h;Inert atmosphere; |
Into a 10-L 3 -necked round-bottom flask purged and maintained with an inert atmosphere of nitrogen Na (164 g, 1.20 equiv) was added in portions to ethanol (5 L). A solution of (C02Et)2 (869 g, 1.00 equiv) and 1-cyclopropylethan-l-one (500 g, 5.94 mol, 1.00 equiv) was added dropwise with stirring at 0-20C. The resulting solution was stirred for 1 h at 20-30C and then for an additional 1 h at 80C. The resulting solution was diluted with 15 L of H20. The pH was adjusted to 2 with hydrochloric acid (12N). The resulting solution was extracted with ethyl acetate and the organic layers combined and washed with NaHCC (sat. aq.). The extract was concentrated under vacuum yielding 820 g (crude) of ethyl 4-cyclopropyl-2,4-dioxobutanoate as yellow oil. TLC (ethyl acetate/petroleum ether =1/5): Rf = 0.5. |
|
With sodium; In ethanol; at 0 - 80℃; for 2h;Inert atmosphere; |
Into a 10-L 3 -necked round-bottom flask purged and maintained with an inert atmosphere of nitrogen Na (164 g, 1.20 equiv) was added in portions to ethanol (5 L). A solution of (C02Et)2 (869 g, 1.00 equiv) and 1-cyclopropylethan-l-one (500 g, 5.94 mol, 1.00 equiv) was added dropwise with stirring at 0-20 C. The resulting solution was stirred for 1 h at 20-30 C and then for an additional 1 h at 80 C. The resulting solution was diluted with 15 L of H20. The pH was adjusted to 2 with hydrochloric acid (12N). The resulting mixture was extracted with ethyl acetate and the organic layers combined and washed with NaHC03 (sat. aq.). The extract was concentrated under vacuum yielding 820 g (crude) of ethyl 4-cyclopropyl-2,4-dioxobutanoate as yellow oil. TLC (ethyl acetate/petroleum ether =1/5): Rf = 0.5. |
|
In ethanol; at 0 - 80℃; for 2h;Inert atmosphere; |
Into a 10-L 3 -necked round-bottom flask purged and maintained with an inert atmosphere of nitrogen Na (164 g, 1.20 equiv) was added in portions to ethanol (5 L). A solution of (C02Et)2 (869 g, 1.00 equiv) and 1-cyclopropylethan-l-one (500 g, 5.94 mol, 1.00 equiv) was added dropwise with stirring at 0-20 C. The resulting solution was stirred for 1 h at 20-30 C and then for an additional 1 h at 80 C. The resulting solution was diluted with 15 L of H20. The pH was adjusted to 2 with hydrochloric acid (12N). The resulting mixture was extracted with ethyl acetate and the organic layers combined and washed with NaHC03 (sat. aq.). The extract was concentrated under vacuum yielding 820 g (crude) of ethyl 4-cyclopropyl-2,4-dioxobutanoate as yellow oil. TLC (ethyl acetate/petroleum ether =1/5): Rf = 0.5. |
| 820 g |
With sodium; In ethanol; at 20 - 80℃; for 2h;Inert atmosphere; |
Into a 10-L 3-necked round-bottom flask purged and maintained with an inert atmosphere of nitrogen Na (164 g, 1.20 equiv) was added in portions to ethanol (5 L). A solution of (C02Et)2 (869 g, 1.00 equiv) and 1-cyclopropylethan-l-one (500 g, 5.94 mol, 1.00 equiv) was added dropwise with stirring at 0-20C. The resulting solution was stirred for 1 h at 20-30C and then for an additional 1 h at 80C. The resulting solution was diluted with 15 L of H20. The pH was adjusted to 2 with hydrochloric acid (12N). The resulting solution was extracted with ethyl acetate and the organic layers combined and washed with NaHC03 (sat. aq.). The extract was concentrated under vacuum yielding 820 g (crude) of ethyl 4-cyclopropyl-2,4-dioxobutanoate as yellow oil. TLC (ethyl acetate/petroleum ether =1/5): Rf = 0.5. |
|
With sodium; In ethanol; at 0 - 80℃; for 2h;Inert atmosphere; |
Into a 10-L 3-necked round-bottom flask purged and maintained with an inert atmosphere of nitrogen Na (164 g, 1.20 equiv) was added in portions to ethanol (5 L). A solution of (CO2Et)2 (869 g, 1.00 equiv) and 1-cyclopropylethan-1-one (500 g, 5.94 mol, 1.00 equiv) was added dropwise with stirring at 0-20oC. The resulting solution was stirred for 1 h at 20-30o C and then for an additional 1 h at 80oC. The resulting solution was diluted with 15 L of H2O. The pH was adjusted to 2 with hydrochloric acid (12N). The resulting solution was extracted with ethyl acetate and the organic layers combined and washed with NaHCO3 (sat. aq.). The extract was concentrated under vacuum yielding 820 g (crude) of ethyl 4-cyclopropyl-2,4-dioxobutanoate as yellow oil. TLC (ethyl acetate/petroleum ether =1/5): Rf = 0.5. |
|
With sodium ethanolate; In ethanol; at -5℃; for 3h; |
General procedure: Sodium ethylate (30.0 g, 0.45 mol) was added into 300 mL of anhydrous ethanol and stirred evenly in ice-salt baths. Then the mixture solution of acetone (14.7 g, 0.3 mol) and diethyl oxalate (46.7 g, 0.32 mol) was added dropwise into sodium ethoxide-ethanol solution to keep the reaction temperature below -5 C for 3 h. The mixture was poured into ice water. After using hydrochloric acid (1 mol/L) to keep the pH around 4, the aqueous phase was extracted with ethyl acetate (EA). The organic layer was washed twice with 150 mL water and dried over Na2SO4, filtered,and concentrated under reduced pressure; 46.5 g (94 %yield, 96 % purity) yellow liquid (1a) was obtained. Without further purification, compound 1a was used in the next reaction. 1b-h were obtained in analogous procedures. |






 Chemistry
Chemistry
 Pharmaceutical Intermediates
Pharmaceutical Intermediates
 Inhibitors/Agonists
Inhibitors/Agonists
 Material Science
Material Science








 For Research Only
For Research Only
 120K+ Compounds
120K+ Compounds
 Competitive Price
Competitive Price
 1-2 Day Shipping
1-2 Day Shipping