|
With oxalyl dichloride; N,N-dimethyl-formamide; In dichloromethane; for 2h; |
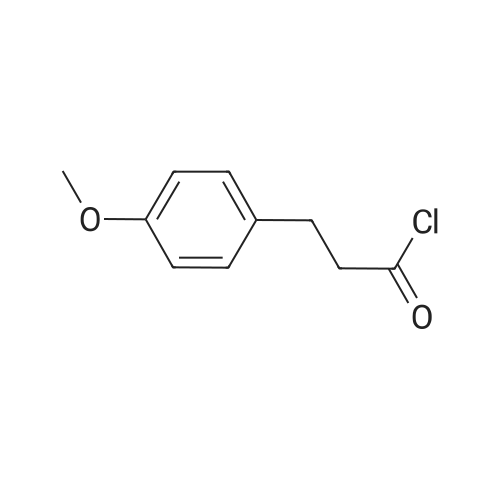
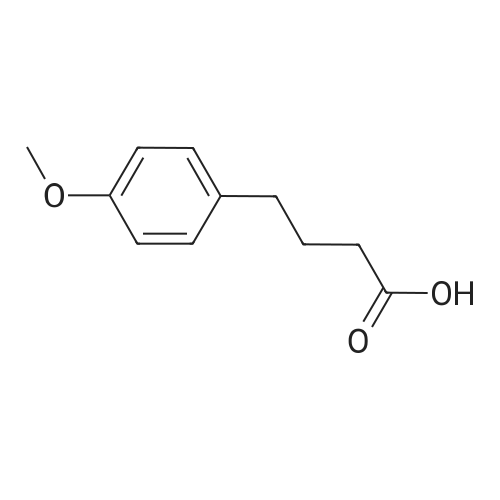
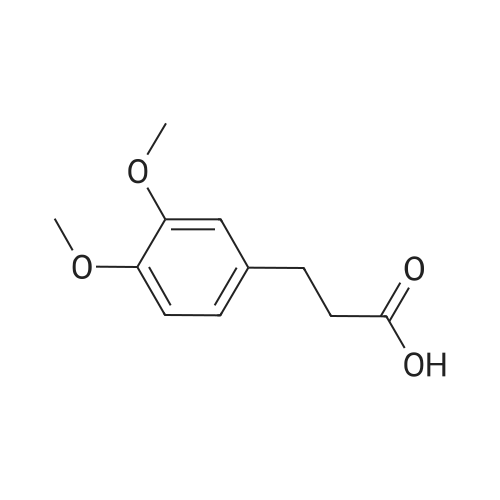
Step 1 Oxalyl chloride (5 eq) was added to a solution of 4-methoxyphenyl)propionic acid (1 eq) in CH2CI2 (0.1 M), and then a catalytic amount of DMF was added to the reaction mixture to initiate the reaction and bubbling occurred. The reaction was complete in 2 hours, and the solution was dried with N2 for one hour to give the acid chloride, which was used immediately in the next step. |
|
With thionyl chloride; In N,N-dimethyl-formamide; toluene; at 75℃; for 3h; |
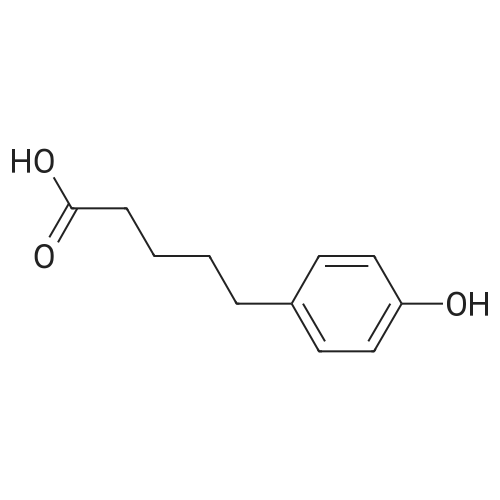
A solution of p-methoxyphenylpropionic acid (12a, 2.99 mmol), SOCl2 (2 mL) and DMF (2 drops) was added to toluene (20 mL) and stirred at 75C for 3 h. The reaction was terminated and the solution was concentrated to dryness under reduced pressure to give p-methoxyphenylpropionyl chloride (13a). (6,2.61 mmol), pyridine (1 mL) in CH2Cl2 (10 mL) was added dropwise with stirring Oxyphenylpropionyl chloride (13a). After completion of the dropwise addition, the reaction was refluxed for 2 h. The reaction was completed, cooled to room temperature, poured into dilute hydrochloric acid, stirred. Extracted with dichloromethane, and washed with saturated brine. The solvent was concentrated to dryness under reduced pressure to give crude product. Column chromatography (petroleum ether / ethyl acetate: 15/1, V / V) gave a white solid in 66.8% |
|
With phosphorus(V) chloride; In dichloromethane;Reflux; |
General procedure: To a solution of phosphorus pentachloride (0.126 g, 0.610 mmol) in dichloromethane (11 mL), phenoxyacetic acid (0.092 g, 0.610 mmol) was added with stirring and the mixture refluxed for 30-40 minutes. After cooling, 4-methyl-1,2,5-oxadiazol-3-amine (0.060 g, 0.610 mmol) was added and the solution refluxed for 2-2.5 hours. The solvent was removed under reduced pressure and the residue quenched with water (50 mL). The solid was collected by vacuum filtration and washed with saturated sodium bicarbonate solution followed by water to afford compound 19 (0.103 g, 73%) |
|
With oxalyl dichloride; N,N-dimethyl-formamide; In dichloromethane; at 20℃; for 5h; |
General procedure: An oven-dried flask was charged with aliphatic carboxylic acid (1.00 equiv) and CH2Cl2 (0.50 M). Three drops of N,N-dimethylformamide (DMF) and oxalyl chloride (1.20 equiv, 1.00 M) in DCM were added dropwise. The reaction mixture was stirred vigorously at room temperature for 5 h and then evaporated DCM and redundant oxalyl chloride under the vacuum. The crude acid chloride was used for the next reaction without any further purification |
|
With oxalyl dichloride; N,N-dimethyl-formamide; at 20℃; for 1h;Inert atmosphere; |
General procedure: Oxalyl chloride (2equiv) was added at room temperature into asolutioncarboxylic acids(1equiv) in anhydrous dichloromethane. Added one drop ofN,N-dimethylformamide and reacted under nitrogen atmosphere for 1h. The solvent was removed under vacuum, yielding a crude product that were stored in a nitrogen atmosphere for subsequent use without further purified. |






 Chemistry
Chemistry
 Pharmaceutical Intermediates
Pharmaceutical Intermediates
 Inhibitors/Agonists
Inhibitors/Agonists
 Material Science
Material Science







 For Research Only
For Research Only
 120K+ Compounds
120K+ Compounds
 Competitive Price
Competitive Price
 1-2 Day Shipping
1-2 Day Shipping