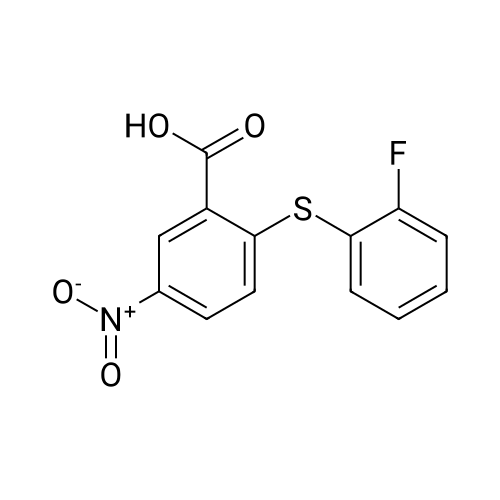
| 56% |
|
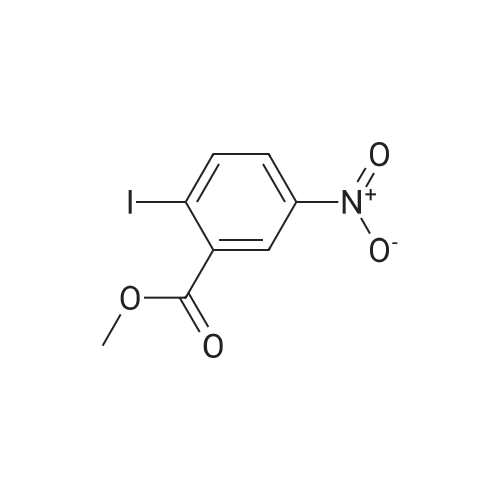
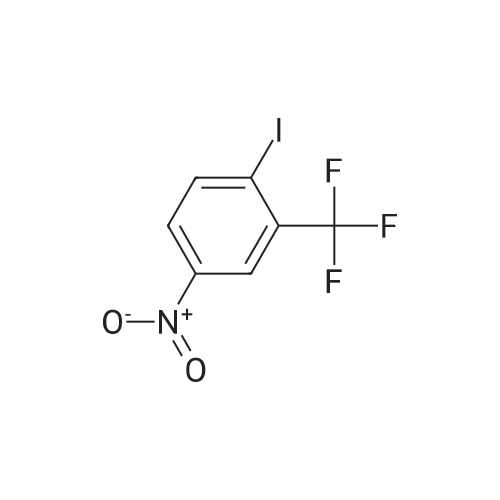
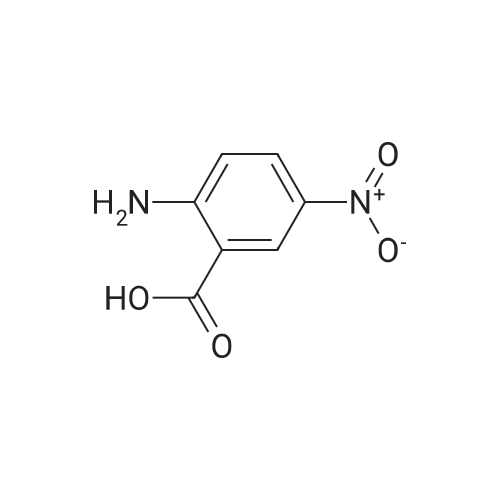
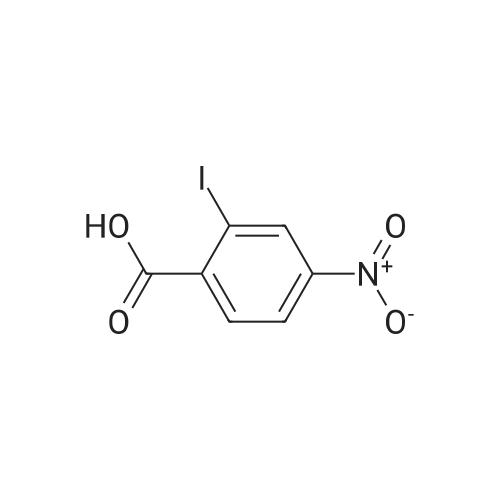
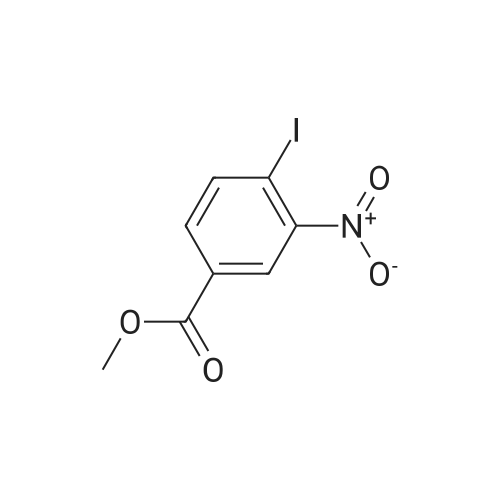
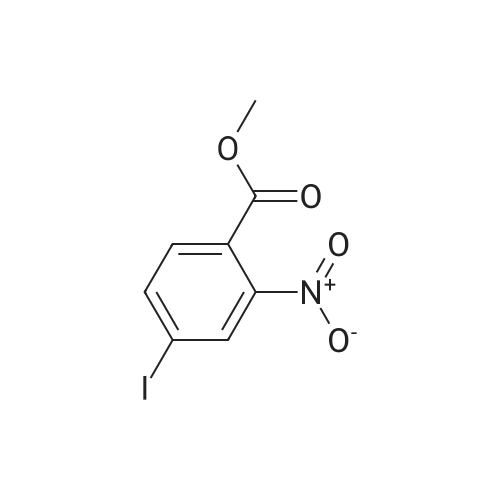
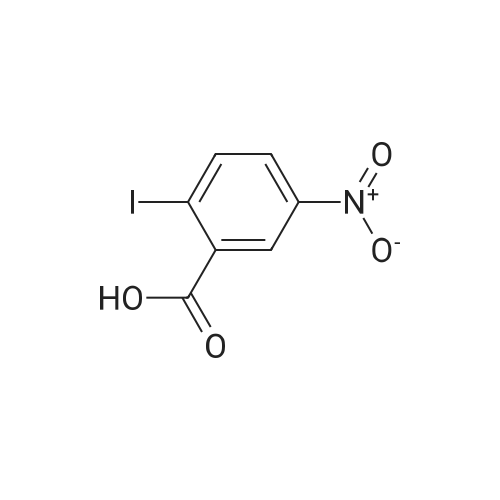
2-Amino-5-nitrobenzoic acid (500 mg, 2.75 mmol) was dissolved in 0.5 M NaOH (5 mL) at 70 C. The resulting solution was allowed to cool to 0 C and the treated with concentrated HCl (1mL). To the mixture was added dropwise a solution of sodium nitrite (196 mg, 2.84 mmol) in water (2.5 mL). After stirring for 0.5 h, a solution of potassium iodide (913 mg, 5.5 mmol) inwater (2.5 mL) was added dropwise to the mixture. The resulting solution was stirred for 1 h and allowed to warm to room temperature. After stirring for 12 h, the precipitate was collected by filtration, washed with water, and dried invacuo. The residue was purified by silica gel column chromatography (eluent: hexane/EtOAc = 1/1) to give 5-nitro-2-iodobenzoic acid [5] (453 mg, 56%) as a pale yellow solid: 1H NMR (400 MHz, CDCl3) delta 8.80 (1H, d, J = 2.8Hz), 8.29 (1H, d, J = 8.7 Hz), 8.03 (1H, dd, J = 8.7, 2.8 Hz). A solution of 5-nitro-2-iodobenzoic acid (293 mg, 1.0mmol) in thionyl chloride (1.3 mL) was heated at reflux with stirring for 2 h. The resulting solution was concentrated under reduced pressure. The remaining thionyl chloride was removed by azeotropic distillation with benzene. The residue was dissolved in anhydrous CH2Cl2 (3.3 mL). To the mixture were added isopropylamine (71 mg, 1.2 mmol)and triethylamine (304 mg, 3.0 mmol) at 0 C under a nitrogen atmosphere. After stirring at room temperature for 21 h,the resulting solution was diluted with EtOAc. The mixture was washed with 10% HCl, saturated aqueous NaHCO3, water, and brine; dried over Na2SO4; filtered; and concentrated under reduced pressure. The residue was purified by recrystallization from hexane and CHCl3 to give 22 (69 mg, 21% in 2 steps) as colorless needles. |
|
|
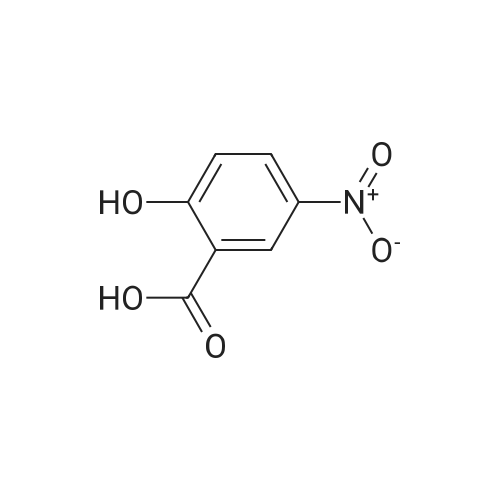
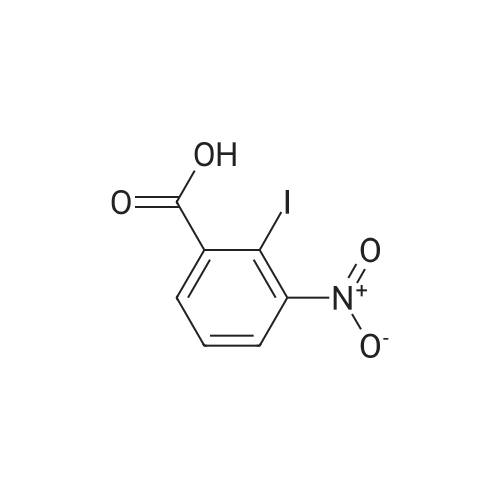
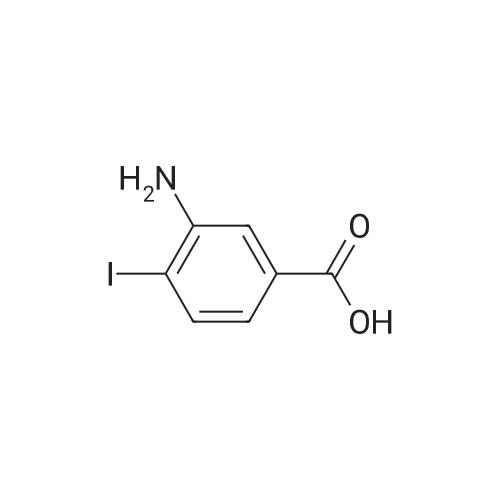
2-Amino-5-nitrobenzoic acid (6d, 1.82 g, 10 mmol) was dissolved in DMSO (50 mL) and 30% H2S04 (50 mL) was added. The resulting mixture was heated for two hours at 50 C. The reaction was cooled to 0 C and a solution of NaN02 (970 mg, 14 mmol) in water (25 mL) was added. The mixture was stirred at 0 C for one hour, whereupon a solution of KI (5.0 g, 30 mmol) in H20 (10 mL) was added and the mixture was stirred for 1 hour at room temperature. Next, another portion of KI (5 g, 30 mmol) in H20 (10 mL) was added and the mixture was stirred for an additional hour. EtOAc (100 mL) was added and the reaction was quenched with saturated aqueous NaHS03 (100 mL). The organic layer was washed with water (2 x 100 mL) and brine (100 mL) and subsequently dried over MgS04. The solvents were evaporated under reduced pressure and the crude product was obtained as yellow solid (12.0 g, 120%). 7d was not further purified and used as a crude in the following reaction. 1H-NMR (400 MHz, CD3OD) delta: 8.54 (d, J = 2.7 Hz, 1H), 8.29 (d, J = 8.6 Hz, 1H), 8.01 (dd, J = 8.7, 2.7 Hz, 1H). 13C-NMR (75 MHz, CD3OD) delta: 168.0, 149.2, 144.1, 139.2, 127.1, 125.8, 103.0, 49.6, 49.3, 49.1, 48.8, 48.5. FT-IR max (cm"1): 2932, 1722, 1588, 151, 1342, 1295, 1022, 1234, 728. HRMS (EI+) m/z calcd for C7H4N04I [M]'+ 292.9185, found 292.9184. |






 Chemistry
Chemistry
 Pharmaceutical Intermediates
Pharmaceutical Intermediates
 Inhibitors/Agonists
Inhibitors/Agonists
 Material Science
Material Science







 For Research Only
For Research Only
 120K+ Compounds
120K+ Compounds
 Competitive Price
Competitive Price
 1-2 Day Shipping
1-2 Day Shipping