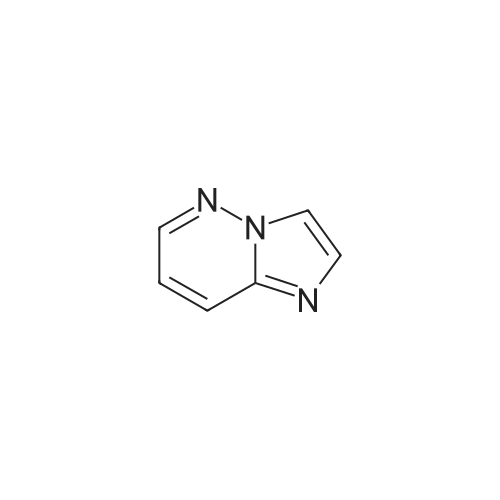
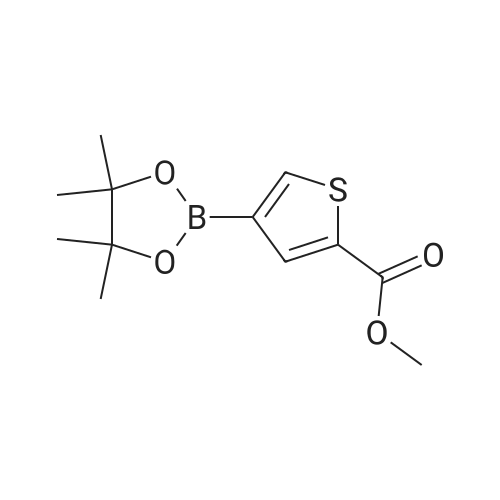
| 74% |
With N-Bromosuccinimide; In chloroform; for 0.5h;Reflux; |
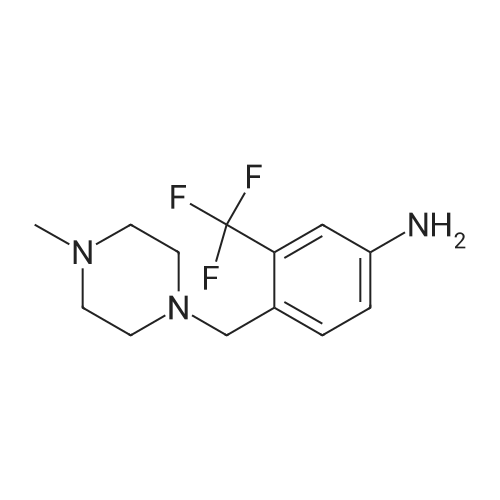
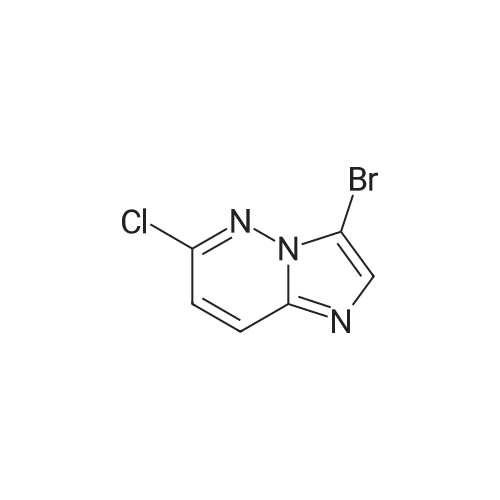
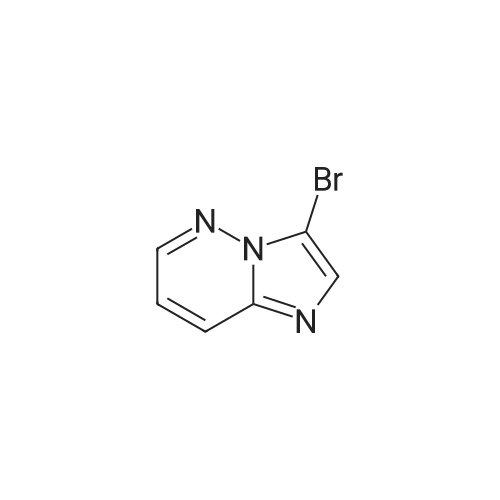
Example 15 -4 Synthesis of 3-bromo-imidazo[1,2-b]pyridazine To imidazo[1,2-b]pyridazine 1*b (10 g) stirring in chloroform (250 ml) was added N-bromosuccinimide (15.7 g) at 5 C. The cooling bath was removed and the reaction was heated to reflux which was maintained for 30 minutes. After leaving to cool overnight the reaction mixture was concentrated under reduced pressure. The residue was redissolved in ethyl acetate (500 ml), washed with potassium carbonate solution (3 x 200 ml) then brine (100 ml). The organic extract was dried with magnesium sulphate and concentrated in vacuo to give 3-bromo-imidazo[1,2-b]pyridazine 2*b, 12.3 g (74%). 1H-NMR (400MHz, DMSO-D6) : delta = 8.68 (1 H, d, 4.4 Hz, ArH), 8.20 (1 H, d, 9.2 Hz, ArH), 7.95 (1 H, s, ArH), 7.33 (1 H, q, 3.4 Hz, ArH). |
| 36% |
With bromine; acetic acid; at 0 - 20℃; for 1h; |
To a stirred solution of imidazo[1,2-bjpyridazine (200 mg, 1.679 mmol) in acetic acid (10 mL) was added bromine (0.2 mL, 3.88 mmol) at 0 C. The reaction mixture was allowed to warm to room temperature and stir for 1 hr. The reaction mixture was neutralized with iN sodium hydroxide, poured into EtOAc (20 mL) and 10% NaHCO3 solution. The layers were separated and the aqueous layer extracted with EtOAC (3x20ml). The combined organic layer was washed with brine, dried over Na2SO4, and concentrated to give 3-bromoimidazo[1,2-bjpyridazine (120 mg, 36%) as light brown solid. ?H NMR (400 MHz, DMSO-d6) oe 8.68 (dd, J=4.52, 1.51 Hz, 1 H) 8.19 (dd, J=9.54, 1.51 Hz, 1 H) 7.94 (s, 1 H) 7.28-7.39 (m, 1 H). |
|
With bromine; In acetic acid; at 20℃; |
EXAMPLE 1; Lambda/-[(l,6i?)-6-Amino-2,2-difluorocyclohexyl]-4-(imidazo[1,2-b]pyridazin-3-yl)-5- methylthiophene-2-carboxamide; 3 -Bromoimidazo [ 1,2-b]pyridazine; Bromine (0.649 mL, 12,6 mmol) was added dropwise to a stirred mixture of Imidazo[1,2- 6]pyridazine (1.0 g, 839 mmol) in acetic acid (42 ml) and the mixture was stirred at room temperature for 1 h. The mixture was neutralized with 1 N sodium hydroxide (100 mL) and solid sodium hydroxide, poured into ethyl acetate and sodium bicarbonate solution, and extracted with ethyl acetate (3 x 200 mL). The combined organics were washed with brine, dried (MgSO,*), and concentrated to afford the title compound. 1H NMR (600 MHz, CD3SOCD3) delta 8.66 (d, 1H); 8.17 (d, 1H); 7.93 (s, 1H); 7.31 (dd, 1H). LRMS (APCI) calc'd for (C6H5BrN3) [M+H]+, 198.0; found 198.0. |
|
With N-Bromosuccinimide; In chloroform; for 2h;Reflux; |
A mixture of imidazo[1,2-b]pyridazine (2.0 g), N-bromosuccinimide (2.94 g), and chloroform (100 mL) was heated under reflux for 2 hr. Upon cooling, the solution was treated with a saturated aqueous solution of sodium carbonate (200 mL) and shaken. The chloroform layer was separated and concentrated to afford the compound 3-bromoimidazo[1,2-b]pyridazine (IIb). |
| 9.9 g |
With N-Bromosuccinimide; In chloroform; for 0.5h;Reflux; |
theImidazo [1,2-b] pyridazin(50mmol) dissolved in Chloroform (50 ml), Bromosuccinimide(55 mmoles) was slowly added into the reaction . at reflux System was stirredfor 30 minutes. After cooling to room temperature, the pH was adjusted to 8-9 usingsaturated aqueous sodium carbonatesolution and extracted with ethyl acetate. The extract was washed with water andsaturated brine. After dried overanhydrous sodium sulfate and concentrated to give 9.9 g of the title compound. |
| 9.9 g |
With N-Bromosuccinimide; In chloroform; for 0.5h;Reflux; |
The imidazo [l, 2_b] pyridazine ¢ .0 g, 50 mmol) was dissolved in chloroform (50 ml), N- bromobutyrate ni imide (NBS) (9.8 g, 55 mmol mol) was slowly added thereto.Was refluxed for 30 minutes.After cooling to room temperature, the PH value was adjusted to 8-9 with saturated sodium carbonate solution, extracted with ethyl acetate.The extract was washed with water, brine, dried over anhydrous sodium sulfate, and concentrated to give the title compound 9.9 g. |
|
With N-Bromosuccinimide; In ethyl acetate; at 95℃; for 2h; |
Was added 3-amino-pyridazine in 250 ml single neck round bottom flask (9.51g, 100mmol), 40% aqueous solution of chloroacetaldehyde (chloroacetaldehyde 190mmol) and 81g of ethyl acetate, Start the magnetic stirrer, the mixture of the reaction flask The reaction was stirred at 85 C for 2 hours. TLC, starting material 3-amino-pyridazine completion of the reaction, was added N- bromosuccinimide (17.80g, 100mmol), 95 under stirring for 2 hours,TLC and GC detection to determine the intermediate imidazo [1,2-b] pyridazine was complete. The reaction mixture was suction filtered, the reaction solution was suction filtered, the filter cake with ethyl acetate: n-hexane = 1: 5 to give the pure product recrystallized from 3-bromo-imidazo [1,2-b] pyridazine, the filtrate with ethyl acetate. The pure product was obtained by recrystallization from ethyl acetate: n-hexane = 1: 5 to give the pure product 3-bromoimidazo [1,2-b] pyridazine. After drying, the yield was calculated 88.98%, purity 99% (HPLC). |






 Chemistry
Chemistry
 Pharmaceutical Intermediates
Pharmaceutical Intermediates
 Inhibitors/Agonists
Inhibitors/Agonists
 Material Science
Material Science







 For Research Only
For Research Only
 120K+ Compounds
120K+ Compounds
 Competitive Price
Competitive Price
 1-2 Day Shipping
1-2 Day Shipping