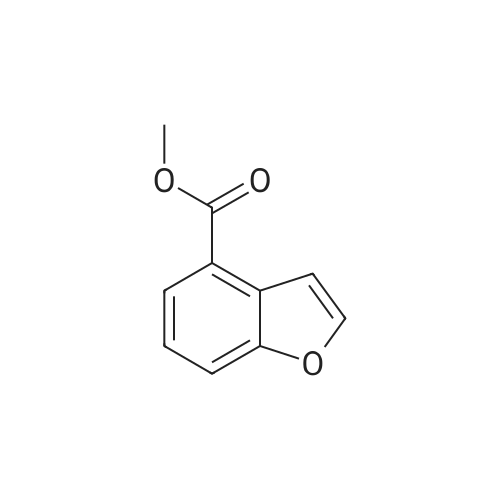
|
With hydrogen;palladium 10% on activated carbon; In ethanol; at 20℃; under 3000.3 Torr; for 16h; |
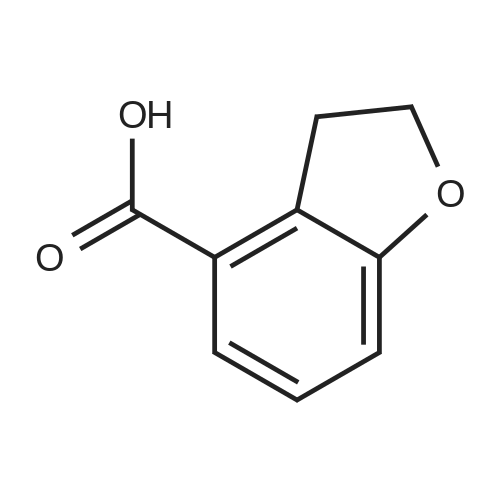
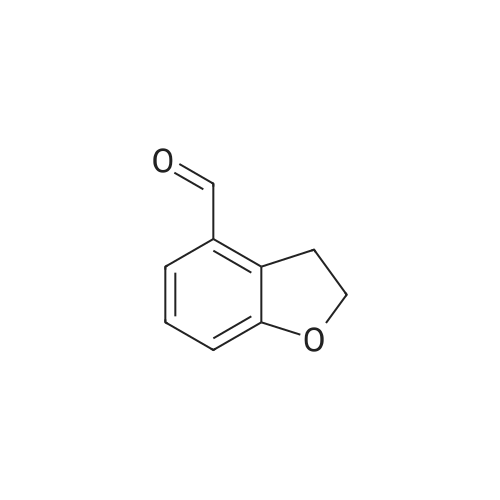
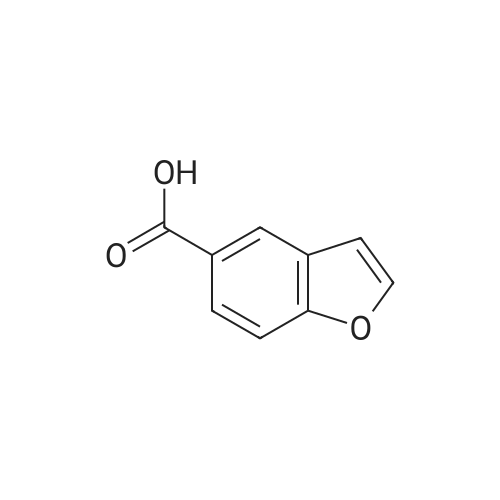
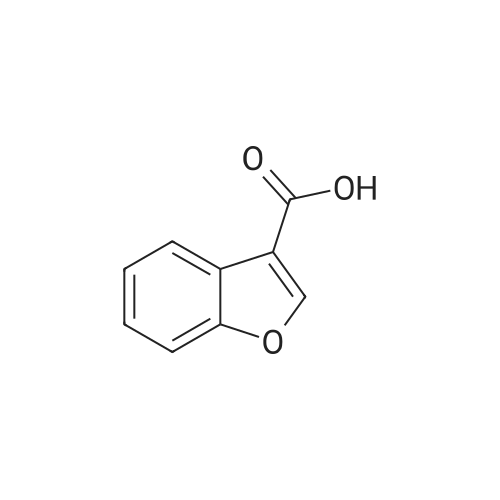
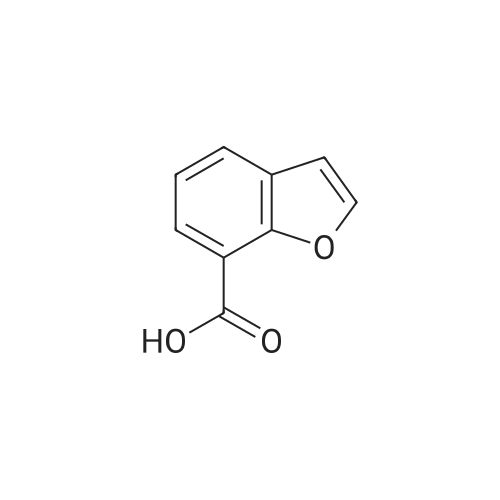
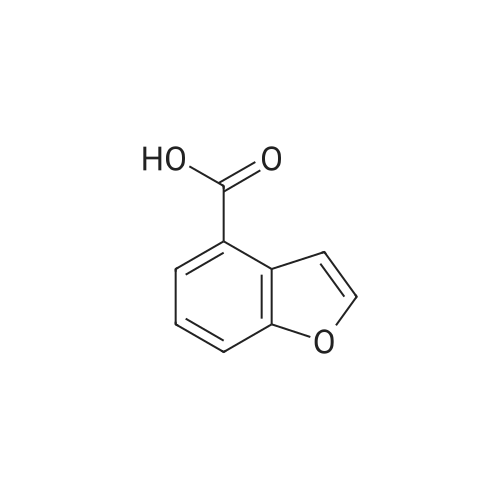
Benzofuran-4-carboxylic acid (30.8 mmol, M.A. Eissenstat et al. J. Med. Chem. 1995, 38, 3094-3105) is added to a suspension of Pd/C (10%, 2.00 g) in EtOH (25 mL). Additional EtOH (75 mL) is added and the mixture is stirred at RT under a hydrogen atmosphere (4 bar) for 16 h. After filtration through celite and removal of the solvents the desired product is obtained which is used without further purification. 1H-NMR (DMSO-de): δ = 3.45 (t, J = 8.79 Hz, 2H); 4.55 (t, J = 8.79 Hz, 2H); 6.99 (d, J = 7.78 Hz, IH); 7.21 (t, J = 7.91 Hz, IH); 7.39 (d, J = 7.78 Hz, IH); 12.9 (bs, IH). |
|
With hydrogen;palladium 10% on activated carbon; In ethanol; at 20℃; under 3000.3 Torr; for 16h; |
Benzofuran-4-carboxylic acid (30.8 mmol, M. A. Eissenstat et al. J. Med. Chem. 1995, 38, 3094-3105) is added to a suspension of Pd/C (10%, 2.00 g) in EtOH (25 mL). Additional EtOH (75 mL) is added and the mixture is stirred at RT under a hydrogen atmosphere (4 bar) for 16 h. After filtration through celite and removal of the solvents the desired product is obtained which is used without further purification. 1H-NMR (DMSO-d6): δ=3.45 (t, J=8.79 Hz, 2H); 4.55 (t, J=8.79 Hz, 2H); 6.99 (d, J=7.78 Hz, 1H); 7.21 (t, J=7.91 Hz, 1H); 7.39 (d, J=7.78 Hz, 1H); 12.9 (bs, 1H). |
|
With hydrogen;palladium 10% on activated carbon; In ethanol; at 20℃; under 3000.3 Torr; for 16h; |
A.2 Synthesis of 2,3-dihydro-<strong>[166599-84-4]benzofuran-4-carboxylic acid</strong> Benzofuran-4-carboxylic acid (30.8 mmol, M. A. Eissenstat et al. J. Med. Chem. 1995, 38, 3094-3105) is added to a suspension of Pd/C (10%, 2.00 g) in EtOH (25 mL). Additional EtOH (75 mL) is added and the mixture is stirred at RT under a hydrogen atmosphere (4 bar) for 16 h. After filtration through celite and removal of the solvents the desired product is obtained which is used without further purification. 1H-NMR (DMSO-d6): δ=3.45 (t, J=8.79 Hz, 2H); 4.55 (t, J=8.79 Hz, 2H); 6.99 (d, J=7.78 Hz, 1H); 7.21 (t, J=7.91 Hz, 1H); 7.39 (d, J=7.78 Hz, 1H); 12.9 (bs, 1H). |
|
With hydrogen;palladium 10% on activated carbon; In acetic acid; under 3102.97 Torr; for 12h; |
Benzofuran-4-carboxylic acid (10.0 g, 61.7 mmol) was hydrogenated (60 psi) in acetic acid (100 mL) over 10% Pd/C (2 g) for 12 hr. The mixture was filtered and the filtrate was diluted with water (500 mL) to give 2,3-dihydro<strong>[166599-84-4]benzofuran-4-carboxylic acid</strong> as a white powder (8.4 g, 83%). A sample was recrystallized from isopropanol to give fine white needles (mp: 185.5-187.5C). |
|
With palladium 10% on activated carbon; hydrogen; In methanol; at 25℃; under 2585.81 Torr; for 12h; |
A third exemplary Intermediate B, Intermediate B-3, may be used to synthesize compounds of formula I wherein Z is O, n is one, X is C(R5)2, - is a single bond and one R3 is chlorine. To a solution of <strong>[166599-84-4]benzofuran-4-carboxylic acid</strong> (900 mg, 5.55 mmol, 1 equiv) in MeOH (9.00 mL) was added palladium on activated carbon (20.0 mg, 555 pmol, 10.0 wt %, 0.10 equiv) under nitrogen. The vessel was evacuated and purged with hydrogen several times. The mixture was stirred at 25 C for 12 h under hydrogen (50.0 psi). The reaction mixture was filtered and the filtrate was concentrated in vacuo to afford 2, 3-dihydro<strong>[166599-84-4]benzofuran-4-carboxylic acid</strong> (750 mg, 3.66 mmol, 65.9% yield, 80.0% purity) as a white solid. 1H NMR (400MHz, DMSO-d6) d = 7.38 (d, J=8.0 Hz, 1H), 7.20 (t, =8.0 Hz, 1H), 6.97 (d, =8.0 Hz, 1H), 4.54 (t, =8.8 Hz, 2H), 3.45 (br t, =8.8 Hz, 2H). |
|
With palladium 10% on activated carbon; hydrogen; In methanol; at 25℃; under 2585.81 Torr; for 12h; |
To a solution of <strong>[166599-84-4]benzofuran-4-carboxylic acid</strong> (900 mg, 5.55 mmol, 1 equiv) in MeOH (9.00 mL) was added palladium on activated carbon (20.0 mg, 555 μmol, 10.0 wt %, 0.10 equiv) under nitrogen. The vessel was evacuated and purged with hydrogen several times. The mixture was stirred at 25 C for 12 h under hydrogen (50.0 psi). The reaction mixture was filtered and the filtrate was concentrated in vacuo to afford 2,3-dihydrobenzofuran-4- carboxylic acid (750 mg, 3.66 mmol, 65.9% yield, 80.0% purity) as a white solid. [0271] 1H NMR (400MHz, DMSO-d6) d = 7.38 (d, J = 8.0 Hz, 1H), 7.20 (t, J = 8.0 Hz, 1H), 6.97 (d, J = 8.0 Hz, 1H), 4.54 (t, J = 8.8 Hz, 2H), 3.45 (br t, J = 8.8 Hz, 2H). |
|
With palladium 10% on activated carbon; hydrogen; In methanol; at 25℃; under 2585.81 Torr; for 12h; |
To a solution of <strong>[166599-84-4]benzofuran-4-carboxylic acid</strong> (900 mg, 5.55 mmol, 1 equiv) in MeOH (9.00 mL) was added palladium on activated carbon (20.0 mg, 555 mol, 10.0 wt %, 0.10 equiv) under nitrogen. The vessel was evacuated and purged with hydrogen several times. The mixture was stirred at 25 C for 12 h under hydrogen (50.0 psi). The reaction mixture was filtered and the filtrate was concentrated in vacuo to afford 2,3-dihydro<strong>[166599-84-4]benzofuran-4-carboxylic acid</strong> (750 mg, 3.66 mmol, 65.9 % yield, 80.0% purity) as a white solid. [0186] 1H NMR (400MHz, DMSO-d6) d = 7.38 (d, J=8.0 Hz, 1H), 7.20 (t, J=8.0 Hz, 1H), 6.97 (d, J=8.0 Hz, 1H), 4.54 (t, J=8.8 Hz, 2H), 3.45 (br t, J=8.8 Hz, 2H). |






 Chemistry
Chemistry
 Pharmaceutical Intermediates
Pharmaceutical Intermediates
 Inhibitors/Agonists
Inhibitors/Agonists
 Material Science
Material Science







 For Research Only
For Research Only
 120K+ Compounds
120K+ Compounds
 Competitive Price
Competitive Price
 1-2 Day Shipping
1-2 Day Shipping