* All experimental methods are cited from the reference, please refer to the original source for details. We do not guarantee the accuracy of the content in the reference.
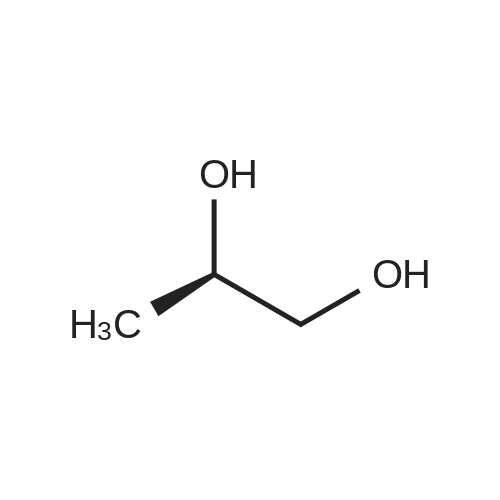
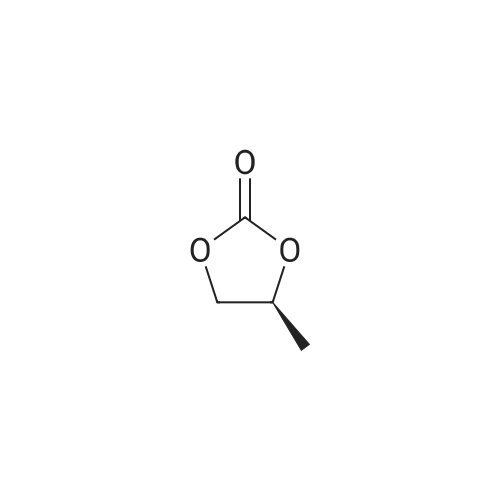
R - Preparation of propylene carbonate,R-1,2-propanediol was added to a glass-lined reactor(38.05 kg, 500Mol),Diethyl carbonate(70.88 kg, 600 mol) and (3.4 kg, 50 mol) of sodium ethoxide,Heated to 105-110 ° C,Reaction for 8 hours,The reaction is terminated,The unreacted diethyl carbonate was distilled off under reduced pressure,Cooled to room temperature,Filter insoluble matter,The filtrate was evaporated to remove the solvent of product R - propylene carbonate 34.24 kg,Purity ≥ 99percentThe yield was 90percent.
81.2%
With sodium methylate In ethanol at 80℃;
Example 1 Preparation of (R)-4-methyl-1,3-dioxolan-2-one To the mixture of diethyl carbonate (380 ml, 15.1 mol) and 200 g of (R)-1,2-propanediol was added 40 ml of denatured ethanol (the solution of 9 g sodium methoxide dissolved in 50 ml of anhydrous ethanol), the resulting solution was heated to 80° C., then ethanol was distilled off slowly. The reaction process was monitored by TLC, after TLC showed that only trace amount of (R)-1,2-propanediol remained or (R)-1,2-propanediol was undetectable, ethanol was distilled under vacuum by water pump at 120° C. until no ethanol dropped out. The residue was distilled under vacuum to give the title compound as a colorless transparent liquid (111 g, 81.2percent yield, purity 97percent by GC)
44.5 g
With sodium ethanolate In ethanol at 80℃;
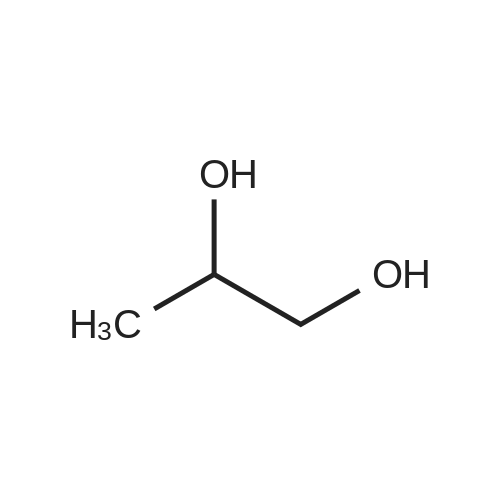
The 1000 ml hydrogenation vessel was evacuated and then passed through nitrogen,Then add 500ml ethanol, 2g5percent Pd / C, 2.5g caustic soda, cool to -10 ,Slowly add 50g of S-glycidol,Then, hydrogen gas (about 2 atmospheres) was passed for 4 hours,To no longer consume hydrogen so far, filtered and concentrated to give 48 g of a colorless oil.
Reference:
[1] Patent: CN105859781, 2016, A, . Location in patent: Paragraph 0011-0012
[2] Patent: US2010/216822, 2010, A1, . Location in patent: Page/Page column 13
[3] Tetrahedron Letters, 1998, vol. 39, # 14, p. 1853 - 1856
[4] Patent: EP1243590, 2002, A2, . Location in patent: Page 9
[5] Patent: CN102977146, 2016, B, . Location in patent: Paragraph 0016; 0021; 0023
[6] Patent: CN108358968, 2018, A, . Location in patent: Paragraph 0073; 0087-0089; 0134-0136; 0142-0144; 0150-0152
[7] Patent: JP2015/164934, 2015, A, . Location in patent: Paragraph 0166; 0167; 0173
2
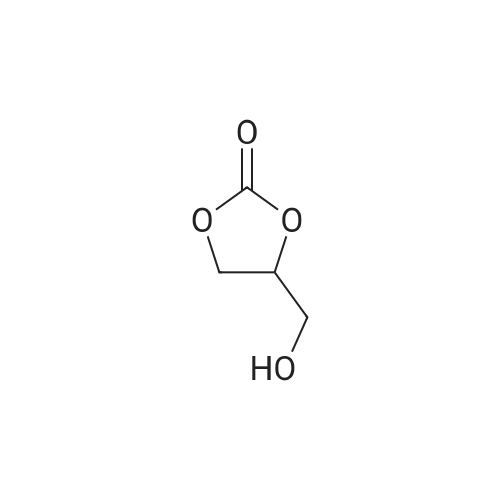
[ 124-38-9 ]
[ 15448-47-2 ]
[ 16606-55-6 ]
Yield
Reaction Conditions
Operation in experiment
99%
at 120℃; for 4 h; Autoclave
General procedure: For a typical reaction of CO2with epoxides, ZnBr2(11.3 mg,0.05 mmol), [BMIM]Br (65 mg, 0.3 mmol) and epoxides (PO: 20 ml,0.286 mol; or ECH: 23.61 g, 0.255 mmol) were charged into a100 ml stainless steel autoclave equipped with a magnetic stirrerin sequence. The 3.0 MPa of CO2was filled into the reaction ves-sel which was then heated to 120C for 4 h. Once the reaction iscompleted, the vessel was cooled to room temperature and thegas was released slowly. Then the liquid phase was analyzed byHP7890A/5975C GC–MS. The unreacted substrate was then dis-tilled out from the mixture, and the product was obtained. Theenantiomeric purity of the mixture was analyzed by HP7890A GCequipped with a chiral DEX120 chromatography column.
Reference:
[1] Journal of Organic Chemistry, 2008, vol. 73, # 20, p. 8039 - 8044
[2] Journal of Molecular Catalysis A: Chemical, 2014, vol. 385, p. 133 - 140
[3] ChemSusChem, 2017, vol. 10, # 15, p. 3025 - 3029
[4] Green Chemistry, 2017, vol. 19, # 16, p. 3769 - 3779
[5] Journal of the American Chemical Society, 2014, vol. 136, # 43, p. 15270 - 15279
[6] Chemical Communications, 2004, # 14, p. 1622 - 1623
[7] Journal of the American Chemical Society, 2001, vol. 123, # 46, p. 11498 - 11499
[8] Advanced Synthesis and Catalysis, 2009, vol. 351, # 9, p. 1325 - 1332
[9] Green Chemistry, 2011, vol. 13, # 3, p. 644 - 650
[10] Inorganic Chemistry, 2012, vol. 51, # 21, p. 12041 - 12052,12
[11] Journal of Organic Chemistry, 2014, vol. 79, # 20, p. 9771 - 9777
[12] ACS Catalysis, 2015, vol. 5, # 11, p. 6773 - 6779
3
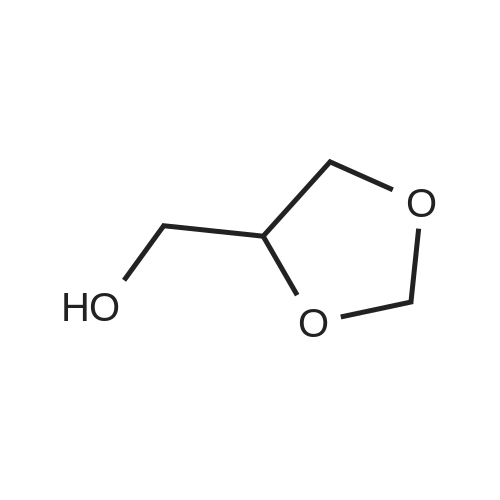
[ 124-38-9 ]
[ 75-56-9 ]
[ 51260-39-0 ]
[ 16606-55-6 ]
Yield
Reaction Conditions
Operation in experiment
34.2%
With C126H91CoN12O10; tetrabutyl-ammonium chloride In dichloromethane at 25℃; for 96 h; Autoclave
General procedure: All reactions were conducted in a 100 mL stainless steel autoclave equipped with a magnetic stir bar, and submerged in anoil bath. The required catalyst, tetrabutylammonium chloride (TBAC) as the co‐catalyst, epoxide, and CH2Cl2 were added tothe reactor in turn. The reactor was then charged with CO2 and vented three times, and finally pressurized with CO2 to 1.0 MPa. The contents were then stirred at room temperature for an established period that depended on the selected substrate and catalyst, after which the reactor was carefully discharged to atmospheric pressure. The yield of cyclic carbonate was determinedby the subtraction method or by comparison betweenthe integral areas obtained by 1H NMR spectroscopy for thecyclic carbonate and epoxide.
31.6%
With C126H91CoN12O10; tetrabutyl-ammonium chloride In dichloromethane at 25℃; for 96 h; Autoclave
General procedure: All reactions were conducted in a 100 mL stainless steel autoclave equipped with a magnetic stir bar, and submerged in anoil bath. The required catalyst, tetrabutylammonium chloride (TBAC) as the co‐catalyst, epoxide, and CH2Cl2 were added tothe reactor in turn. The reactor was then charged with CO2 and vented three times, and finally pressurized with CO2 to 1.0 MPa. The contents were then stirred at room temperature for an established period that depended on the selected substrate and catalyst, after which the reactor was carefully discharged to atmospheric pressure. The yield of cyclic carbonate was determinedby the subtraction method or by comparison betweenthe integral areas obtained by 1H NMR spectroscopy for thecyclic carbonate and epoxide.
38.8 % ee
at 25℃; for 25 h; Autoclave
General procedure: A solution of freshly prepared SalenCo(III)Y (0.1 mmol) andepoxide (100 mmol) was introduced into a 100 mL stainless-steelautoclave, which was purged three times and charged with CO2to 1.2 MPa. The reaction mixture was stirred at room temperature.When the pressure of reactor was fall down to a presetting value, itwas then vented carefully. After removing the excess epoxide, theresidue was weighed to measure the conversion of epoxide, chiralcyclic carbonate (R = Me, Et, CH2Cl) was distilled under vacuum as asa colorless liquid or it (R = Ph, PhOCH2) was obtained by columnchromatography through a short silica–gel column to yield whitesolid product (ethyl acetate/petroleum ether = 5:1).
Reference:
[1] Dalton Transactions, 2013, vol. 42, # 27, p. 9930 - 9937
[2] Chinese Journal of Catalysis, 2018, vol. 39, # 5, p. 997 - 1003
[3] Chinese Journal of Catalysis, 2018, vol. 39, # 5, p. 997 - 1003
[4] Journal of the American Chemical Society, 2004, vol. 126, # 12, p. 3732 - 3733
[5] Chemical Communications, 2004, # 14, p. 1622 - 1623
[6] Tetrahedron Letters, 2007, vol. 48, # 2, p. 297 - 300
[7] Tetrahedron Letters, 2008, vol. 49, # 46, p. 6589 - 6592
[8] Advanced Synthesis and Catalysis, 2009, vol. 351, # 9, p. 1325 - 1332
[9] Advanced Synthesis and Catalysis, 2009, vol. 351, # 9, p. 1325 - 1332
[10] Green Chemistry, 2009, vol. 11, # 7, p. 935 - 938
[11] Green Chemistry, 2009, vol. 11, # 7, p. 935 - 938
[12] Science China Chemistry, 2011, vol. 54, # 7, p. 1044 - 1050
[13] Science China Chemistry, 2011, vol. 54, # 7, p. 1044 - 1050
[14] Catalysis Science and Technology, 2013, vol. 3, # 10, p. 2661 - 2667
[15] Journal of Molecular Catalysis A: Chemical, 2016, vol. 411, p. 34 - 39
[16] RSC Advances, 2016, vol. 6, # 4, p. 3243 - 3249
[17] Chemical Communications, 2017, vol. 53, # 79, p. 10930 - 10933
[18] Chemical Communications, 2017, vol. 53, # 79, p. 10930 - 10933
[19] Chinese Journal of Catalysis, 2018, vol. 39, # 5, p. 997 - 1003
4
[ 124-38-9 ]
[ 75-56-9 ]
[ 51260-39-0 ]
[ 15448-47-2 ]
[ 16606-55-6 ]
Yield
Reaction Conditions
Operation in experiment
66.7 % ee
at 25℃; for 8 h; Autoclave
General procedure: Catalyst of (S,R,R,S)-ZSS-2b (631 mg, 0.1 mmol), cocatalyst of tetrabutylammonium fluoride (TBAF, 0.0522 g, 0.2 mmol), and racemic propylene oxide (PO, 7 mL, 100 mmol) were introduced into a 100-mL stainless steel autoclave to form a brown solution. The reactor was purged thrice with carbon dioxide, and charged it to 0.8 MPa. Then, the asymmetric catalytic reaction took place at room temperature. After some time, the pressure in the reactor decreased to an expected value, and it was vented to terminate the reaction. After removing the unreacted epoxide, chiral cyclic carbonate (R=Me, Et, CH2Cl), weighed to calculate the yield of cyclic carbonate, was distilled under vacuum as a colorless liquid or it (R=Ph, PhOCH2) was obtained by column chromatography through a short silica gel column (ethyl acetate:petroleum ether = 5:1).
General procedure: Catalyst of (S,R,R,S)-ZSS-2b (631 mg, 0.1 mmol), cocatalyst of tetrabutylammonium fluoride (TBAF, 0.0522 g, 0.2 mmol), and racemic propylene oxide (PO, 7 mL, 100 mmol) were introduced into a 100-mL stainless steel autoclave to form a brown solution. The reactor was purged thrice with carbon dioxide, and charged it to 0.8 MPa. Then, the asymmetric catalytic reaction took place at room temperature. After some time, the pressure in the reactor decreased to an expected value, and it was vented to terminate the reaction. After removing the unreacted epoxide, chiral cyclic carbonate (R=Me, Et, CH2Cl), weighed to calculate the yield of cyclic carbonate, was distilled under vacuum as a colorless liquid or it (R=Ph, PhOCH2) was obtained by column chromatography through a short silica gel column (ethyl acetate:petroleum ether = 5:1).
Reference:
[1] Synthesis, 2009, # 8, p. 1403 - 1404
7
[ 128765-74-2 ]
[ 1015235-29-6 ]
[ 1015235-28-5 ]
[ 16606-55-6 ]
Yield
Reaction Conditions
Operation in experiment
99.67 % ee
With sodium hydroxide In water at 20℃; for 25.5 h; Aqueous phosphate buffer; D-glucose
Example 1 Biocatalytic reduction of 0- (methoxycarbonyl) -hydroxyacetone, 2a :4aIn a Titrino reaction vessel, the whole-cell catalyst of type E. coli DSM14459, containing an (R) -alcohol dehydrogenase from L. kefir and a glucose dehydrogenase from T. acidophilum (for production of the biocatalyst, see WO2005121350) , at a cell concentration of 55 g moist biomass / L, D-glucose (1.5 equivalents relative to the molar amount of ketone used) and 25 mmol 0-(methyloxycarbonyl) -hydroxyacetone, 2a, (corresponding to a substrate concentration of 0.5M) are added to 30 mL of an aqueous phosphate buffer (0.026 M; adjusted to pH 7.0) and the volume is topped up to 50 mL with water. The reaction mixture is stirred for a reaction time of 25.5 hours at room temperature, maintaining constant pH at -6.5 by adding sodium hydroxide solution (5M NaOH). After a reaction time of 25.5 hours, conversion of >95percent is determined (according to the consumption of sodium hydroxide solution and GC chromatography) . Processing is carried out by lowering the pH value to <3 with concentrated hydrochloric acid and addition of 3.75 g of the filter aid Celite Hyflo <n="25"/>Supercel to the reaction mixture, followed by filtration with application of vacuum. The filter cake is washed 4 times with 50 mL MTBE and the aqueous phase is extracted correspondingly with the three organic MTBE fractions obtained. The solvent is removed from the combined organic phases after drying over magnesium sulphate, yielding as raw product the optically active alcohol 3a at a yield of 50percent (of which 12.7 mol.percent is rearranged to give the regioisomeric alcohol 4a and 63.6 mol.percent has already been cyclized to the desired (R) -propylene carbonate 1) . The enantioselectivity of the reaction is 99.67percent ee .
With sodium hydroxide In water at 20℃; for 25.5 h; Aqueous phosphate buffer; D-glucose
Example 2_Biocatalytic reduction of 0- (ethoxycarbonyl) - hydroxyacetone, 2b:4bIn a Titrino reaction vessel, the whole-cell catalyst of type E. coli DSM14459, containing an (R) -alcohol dehydrogenase from L. kefir and a glucose dehydrogenase from T. acidophilum (for production of the biocatalyst, see WO2005121350) , at a cell concentration of 51 g moist biomass / L, D-glucose (1.5 equivalents relative to the molar amount of ketone used) and 25 mmol 0- (ethyloxycarbonyl) -hydroxyacetone, 2b, (corresponding to a substrate concentration of 0.5M) are added to 30 mL of an aqueous phosphate buffer (0.026 M; adjusted to pH 7.0) and the volume is topped up to 50 mL with water. The reaction mixture is stirred for a reaction time of 25.5 hours at room temperature, maintaining constant pH at -6.5 by adding sodium hydroxide solution (5M NaOH) . After a reaction time of 25.5 hours, conversion of >95percent is determined (according to the consumption of sodium hydroxide solution and GC chromatography) . Processing is carried out by lowering the pH value to <3 with concentrated hydrochloric acid and addition of 3.75 g of the filter aid Celite Hyflo Supercel to the reaction mixture, followed by filtration with application of vacuum. The filter cake is washed 4 times with 50 mL MTBE and the aqueous phase <n="27"/>is extracted correspondingly with the three organicMTBE fractions obtained. The solvent is removed from the combined organic phases after drying over magnesium sulphate, yielding as raw product the optically active alcohol 3b at a yield of 71percent (of which 37.3 mol.percent is rearranged to give the regioisomeric alcohol 4a and18.6 mol.percent has already been cyclized to the desired(R) -propylene carbonate 1) . The enantioselectivity of the reaction is 99.34percent ee
Example 4_Synthesis of (R) -propylene carbonate 1 by cyclization of the raw product from Example 2:HO.XH-,4b0.525 g of the optically active alcohol 3b obtained as raw product according to example 2 (which has partially been rearranged to give the regioisomeric alcohol 4b or has already been cyclized to the desired (R) -propylene carbonate according to the proportions stated in mol.percent in example 2) is absorbed in 10 mL ethyl acetate, and p-toluenesulphonic acid (96 mg) is added. The reaction mixture is heated for 6 hours at a reaction temperature of 600C. The desired (R) -propylene carbonate 1 is obtained in a proportion of -80percent (relative to the molar quantity of substrate used from example 2) and with an enantioselectivity of 99.18percent ee






 Chemistry
Chemistry
 Pharmaceutical Intermediates
Pharmaceutical Intermediates
 Inhibitors/Agonists
Inhibitors/Agonists
 Material Science
Material Science







 For Research Only
For Research Only
 120K+ Compounds
120K+ Compounds
 Competitive Price
Competitive Price
 1-2 Day Shipping
1-2 Day Shipping