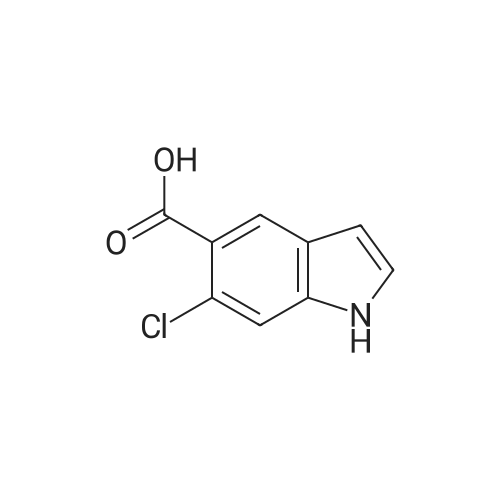
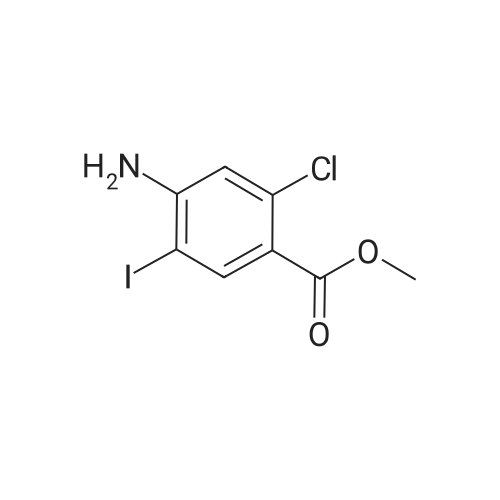
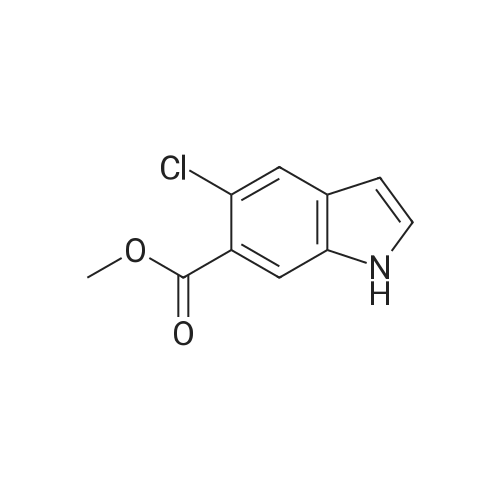
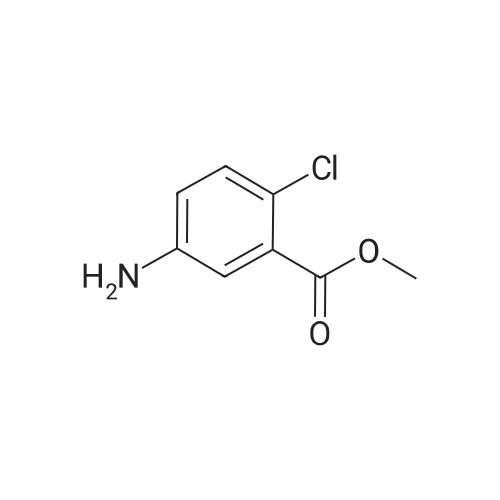
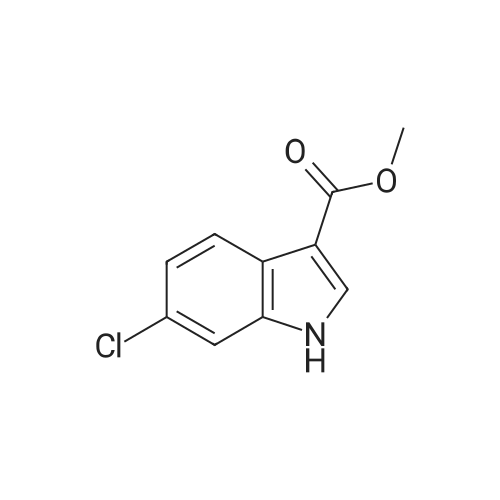
| 83.2 - 92.0% |
With tetrabutyl ammonium fluoride; In acetonitrile; at 20 - 80℃; for 7h;Product distribution / selectivity; |
EXAMPLE 3; Preparation of 6-Chloro-5-methoxycarbonyl Indole; A solution of tetrabutylammonium fluoride (TBAF) in acetonitrile was prepared by dissolving 372 kg of TBAF trihydrate in acetonitrile (720 kg) with stirring for at least one hour. 2-Chloro-4-acetamido-5-(2-trimethylsilylethynyl)-benzoic acid methyl ester (ca. 300 kg) was dissolved in acetonitrile (1168 kg) at 20-30 C. The TBAF/acetonitrile solution was added to this solution with stirring, while the temperature was maintained at less than 35 C. The TBAF solution was rinsed in with another portion (49 kg) of acetonitrile. This mixture was heated to 78-80 C. for 7 hours or until less than 1% starting material remained as judged by HPLC, and the product accounted for at least 93% of the material by HPLC. The mixture was then vacuum distilled at a temperature not exceeding 35 C. to a volume of about 1200 L. It was then split into two batches, each of which was combined with about 1900 L water at 15-25 C. for one hour to precipitate the product. About half of each batch was spun down in a peeler centrifuge and rinsed with water (198 L); then the other half of each batch was processed similarly. When all of both batches had been processed, the collected off-white to pale brown solids were dried at 35-40 C. to provide a 91-92% yield of material that was 95-97% pure as judged by HPLC. Laboratory work suggests that this yield may be improved by reducing the volume of acetonitrile present, as by vacuum distillation, prior to the aqueous quench, and by increasing the volume of water used for the quench. This has not been tested on large scale, however. The above examples are simplified descriptions of representative scale-up and pilot plant runs for each of these specific reactions. More detailed descriptions of these examples, including comparison of multiple batches up to pilot plant scale, are presented in the following examples.; EXAMPLE 6 Prior to Campaign S3, tetrabutylammonium fluoride (TBAF) and Stage 4 were charged to the reactor as solids before adding solvent. In Campaign S2, this led to an exothermic reaction between the solids with potential for serious safety consequences. For Campaign S3, TBAF was added successfully as a THF solution. In Campaign S4, Stage 5 was isolated for the first time as a solid to allow the possibility of registering it as a ?cGMP Starting Material?. The reaction solvent was changed from THF to acetonitrile and the product was isolated by distilling off excess solvent and adding to a large volume of water. The modified process worked well and yielded Stage 5 product with satisfactory quality. In Campaign S5, the batch size (Stage 4 input) was increased by a factor of 2.18 and the initial acetonitrile charge was increased from 1.9 to 5.5 volumes to compensate for the very high minimum stirring volume in the reaction vessel. The excess acetonitrile was removed by vacuum distillation prior to quenching the reaction mixture. The process employed in Campaign S6 was virtually unchanged from S5 apart from a 31% increase in batch size. This increase permitted a smaller volume of acetonitrile to be used for the reaction. However, due to volume constraints it was necessary to quench the batch in two halves. An additional change introduced for both S5 and S6 was the isolation of the product on a centrifuge in place of a vat filter. This generally resulted in better de-liquoring of the wet-cake leading to improved purity and shorter drying times. Comparison of Yield and Purity for Campaigns S3 to S6. TABLE ONE Stage 5 Yield and Purity Data for Campaigns S4 to S6 Campaign Stage 5 Campaign Assay- HPLC Physical based Purity Stage 5 Batch Stage 4 Input Stage 5 Output Yield Yield (Area Campaign Number (kg) (kg) (%) (%) %) S4 80011358A, B 99.0 58.2 90.8 83.2 94.7 S5 800125330 202.2 115.4 90.6 89.1 96.4 800125780 210.5 126.6 96.3 S6 80013075A, B 266.2 157.2 92.0 91.4 95.4 80013076A, B 273.4 164.2 97.0 The data show that physical yield has remained virtually unchanged since Campaign S4. However, Campaigns S5 and S6 showed a dramatic improvement in assay-based yield. The reasons for this are not fully understood. Certainly, the material isolated in S5 and S6 had a much higher assay (>93% w/w) than the S4 material (84.8% w/w). This was attributed to isolation on a centrifuge, giving more efficient liquor removal and thus reduced retention of impurities. Similarly, the Stage 4 input for S5 and S6 was purer (>93.8% w/w) than that used in S4. These facts in themselves do not explain the dramatic increase in assay-based yield. However, it is possible that a purer Stage 4 input may have resulted in a better crystallization and thus a more efficient product recovery. The Stage 5 product generated in Campaigns S5 and S6 had a higher area % purity than that from S5. Also, as noted above, the % w/w assay was much higher for the S5 and S6 material. Both these observations are consistent with the use of centrifugation in S5 and S6 and the consequent imp... |






 Chemistry
Chemistry
 Pharmaceutical Intermediates
Pharmaceutical Intermediates
 Inhibitors/Agonists
Inhibitors/Agonists
 Material Science
Material Science







 For Research Only
For Research Only
 120K+ Compounds
120K+ Compounds
 Competitive Price
Competitive Price
 1-2 Day Shipping
1-2 Day Shipping