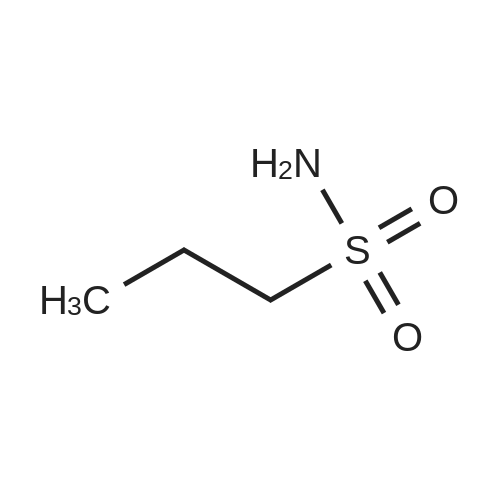
| 100% |
With ammonia; In tetrahydrofuran; at 0 - 20℃;Product distribution / selectivity; |
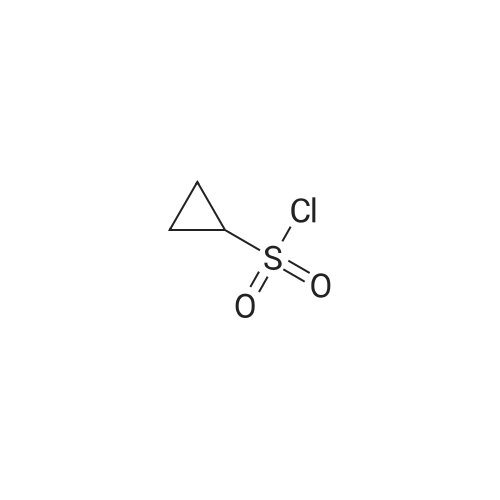
To a solution of 100 mL of THF cooled to 0 C. was bubbled in gaseous ammonia until saturation was reached. To this solution was added a solution of 5 g (28.45 mmol) of cyclopropylsulfonyl chloride (purchased from Array Biopharma) in 50 mL of THF, the solution warmed to rt overnite and stirred one additional day. The mixture was concentrated until 1-2 mL of solvent remained, applied on to 30 g plug of SiO2 (eluted with 30% to 60% EtOAc/Hexanes) to afford 3.45 g (100%) of cyclopropyl sulfonamide as a white solid. 1H NMR (Methanol-d4) δ 0.94-1.07 (m, 4H), 2.52-2.60 (m, 1H); 13C NMR (methanol-d4) δ 5.92, 33.01. |
| 100% |
With ammonia; In tetrahydrofuran; at 0 - 20℃; |
To a solution of 100 mL of THF cooled to 0 C was bubbled in gaseous ammonia until saturation was reached. To this solution was added a solution of 5 g(28. 45 mmol) of cyclopropylsulfonyl chloride (purchased from ArrayBiopharma) in 50mL of THF, the solution warmed to rtovernite and stirred one additional day. The mixture was concentrated until 1-2mL of solvent remained, applied onto 30 g plug ofSi02 (eluted with 30% to 60% EtOAc/Hexanes) to afford 3.45g (100%) of cyclopropyl sulfonamide as a whitesolid.'H NMR (Methanol-d4) 0.94-1. 07 (m, 4H), 2.52-2. 60 (m,1H); 13C NMR(methanol-d4)8 5.92, 33.01. |
| 100% |
With ammonia; In tetrahydrofuran; at 0 - 20℃; |
Method A: To a solution of 100 mL of TUF cooled to 0 C. was bubbled in gaseous ammonia until saturation was reached. To this solution was added a solution of 5 g (28.45 mmol) of cyclopropylsulfonyl chloride (purchased from Array Biopharma) in 50 mL of THF, the solution warmed to rt overnite and stirred one additional day. The mixture was concentrated until 1-2 mL of solvent remained, applied onto 30 g plug of SiO2 (eluted with 30% to 60% EtOAc/Hexanes) to afford 3.45 g (100%) of cyclopropylsulfonamide as a white solid. |
| 100% |
With ammonia; In tetrahydrofuran; at 0 - 20℃;Product distribution / selectivity; |
To a solution of 100 mL of THF cooled to 0 C was bubbled in gaseous ammonia until saturation was reached. To this solution was added a solution of 5 g (28.45 mmol) of cyclopropylsulfonyl chloride (purchased from Array Biopharma) in 50 mL of THF, the solution warmed to rt overnite and stirred one additional day. The mixture was concentrated until 1-2 mL of solvent remained, applied onto 30 g plug of Si02 (eluted with 30% to 60% EtOAc/Hexanes) to afford 3. 45g (100%) of cyclopropyl sulfonamide as a white solid NMR (Methanol-d4) 8 0.94-1. 07 (m, 4H), 2.52-2. 60 (m, 1H) ; 13C NMR (methanol-d4) 8 5.92, 33.01. |
| 100% |
With ammonia; In tetrahydrofuran; at 20℃;Product distribution / selectivity; |
Method A:; O NH3 ( sat ) THF O^ ^ -S I l -Cl ? n _ h I ^ S -NHO 0 0C to rt OTo a solution of 100 mL of THF cooled to 0 0C was bubbled in gaseous ammonia until saturation was reached. To this solution was added a solution of 5 g (28.45 mmol) of cyclopropylsulfonyl chloride (purchased from Array Biopharma) in 50 mL of THF, the solution warmed to rt overnite and stirred one additional day. The mixture was concentrated until 1-2 mL of solvent remained, applied on to 30 g plug of SiO2 (eluted with 30% to 60% EtOAc/Hexanes) to afford 3.45g (100%) of cyclopropyl sulfonamide as a white solid. 1H NMR (Methanol^) δ 0.94-1.07 (m, 4H), 2.52-2.60 (m, IH); 13C NMR (methanol^) δ 5.92, 33.01. |
| 100% |
With ammonia; In tetrahydrofuran; at 0 - 20℃;Product distribution / selectivity; |
To a solution of 100 mL of THF cooled to 0 C. was bubbled in gaseous ammonia until saturation was reached. To this solution was added a solution of 5 g (28.45 mmol) of cyclopropylsulfonyl chloride (purchased from Array Biopharma) in 50 mL of THF, the solution warmed to rt overnite and stirred one additional day. The mixture was concentrated until 1-2 mL of solvent remained, applied on to 30 g plug of SiO2 (eluted with 30% to 60% EtOAc/Hexanes) to afford 3.45 g (100%) of cyclopropyl sulfonamide as a white solid. 1H NMR (Methanol-d4) δ 0.94-1.07 (m, 4H), 2.52-2.60 (m, 1H); 13C NMR (methanol-d4) δ 5.92, 33.01. |
| 100% |
With ammonia; In tetrahydrofuran; at 0 - 20℃; for 24h;Product distribution / selectivity; |
Method A To a solution of 100 mL of THF cooled to 0 C. was bubbled in gaseous ammonia until saturation was reached. To this solution was added a solution of 5 g (28.45 mmol) of cyclopropylsulfonyl chloride (purchased from Array Biopharma) in 50 mL of THF, the solution warmed to it overnite and stirred one additional day. The mixture was concentrated until 1-2 mL of solvent remained, applied on to 30 g plug of SiO2 (eluted with 30% to 60% EtOAc/Hexanes) to afford 3.45 g (100%) of cyclopropyl sulfonamide as a white solid. 1H NMR (Methanol-d4) δ 0.94-1.07 (m, 4H), 2.52-2.60 (m, 1H); 13C NMR (methanol-d4) δ 5.92, 33.01. |
| 100% |
With ammonia; In tetrahydrofuran; at 0 - 20℃;Product distribution / selectivity; |
To a solution of 100 mL of THF cooled to 0 C. was bubbled in gaseous ammonia until saturation was reached. To this solution was added a solution of 5 g (28.45 mmol) of cyclopropylsulfonyl chloride (purchased from Array Biopharma) in 50 mL of THF, the solution warmed to rt overnight and stirred one additional day. The mixture was concentrated until 1-2 mL of solvent remained, applied on to 30 g plug of SiO2 (eluted with 30% to 60% EtOAc/Hexanes) to afford 3.45 g (100%) of cyclopropyl sulfonamide as a white solid. 1H NMR (Methanol-d4) δ 0.94-1.07 (m, 4H), 2.52-2.60 (m, 1H); 13C NMR (methanol-d4) δ 5.92, 33.01. |
| 100% |
With ammonia; In tetrahydrofuran; at 0 - 20℃; |
To a solution of 100 mL of THF cooled to 0 C. was bubbled in gaseous ammonia until saturation was reached. To this solution was added a solution of 5 g (28.45 mmol) of cyclopropylsulfonyl chloride (purchased from Array Biopharma) in 50 mL of THF, the solution warmed to rt overnite and stirred one additional day. The mixture was concentrated until 1-2 mL of solvent remained, applied on to 30 g plug of SiO2 (eluted with 30% to 60% EtOAc/Hexanes) to afford 3.45 g (100%) of cyclopropyl sulfonamide as a white solid. 1H NMR (Methanol-d4) δ 0.94-1.07 (m, 4H), 2.52-2.60 (m, 1H); 13C NMR (methanol-d4) δ 5.92, 33.01. |
| 100% |
With ammonia; In tetrahydrofuran; at 0 - 20℃;Product distribution / selectivity; |
Method 2 To a solution of 100 mL of THF cooled to 0 C. was bubbled in gaseous ammonia until saturation was reached. To this solution was added a solution of 5 g (28.45 mmol) of cyclopropylsulfonyl chloride (purchased from Array Biopharma) in 50 mL of THF. The solution was warmed to room temperature overnight and stirred one additional day. The mixture was concentrated until 1-2 mL of solvent remained and poured onto a 30 g plug of SiO2 (eluted with 30% to 60% ethyl acetate/hexanes) to provide 3.45 g (100%) of cyclopropylsulfonamide as a white solid. 1H NMR (methanol-d4) δ 0.94-1.07 (m, 4H), 2.52-2.60 (m, 1H); 13C NMR (methanol-d4) δ 5.92, 33.01. |
| 100% |
With ammonia; In tetrahydrofuran; at 0 - 20℃;Product distribution / selectivity; |
To a solution of 100 mL of THF cooled to 0 0C was bubbled in gaseous ammonia until saturation was reached. To this solution was added a solution of 5 g (28.45 mmol) of cyclopropylsulfonyl chloride (purchased from Array Biopharma) in 50 mL of THF, the solution warmed to rt overnite and stirred one additional day. The mixture was concentrated until 1-2 mL of solvent remained, applied on to 30 g plug of SiO2 (eluted with 30% to 60% EtOAc/Hexanes) to afford 3.45g (100%) of cyclopropyl sulfonamide as a white solid. 1H NMR (Methanol^) δ 0.94-1.07 (m, 4H), 2.52-2.60 (m, IH); 13C NMR (methanol^) δ 5.92, 33.01. |
| 95% |
With ammonia; In dichloromethane; at -78 - 20℃; for 3.25h; |
Example 27Synthesis of new sulfonamides[0529] Preparation of compound 3: compound 1 (1Og, 71mmol) was dissolved in dry DCM (150ml) under nitrogen. The resulting solution was bubbled through NH3 for 15 minutes at -78 -C, then it was allowed to rise to room temperature and stirred for 3 h. Before filtration, the Filtrate was concentrated to provide compound 2 as white solid 8.2 g (yield |
| > 90% |
With ammonia; In 1,4-dioxane; at 20℃; for 72h; |
Cyclopropylsulfonyl chloride (5 g, 35.56 mmol) was dissolved in 0.5 M ammonia in dioxane (200 ml, 100 mmol) at RT. The reaction was stirred at RT for 3 days. The large amount of precipitation was filtered and discarded. The clear filtrate was evaporated in vacuo and the white residue was dried on vacuum for 24 hours to give the cyclopropylsulfonamide (>90%). 1H-NMR (500 MHz, CD3Cl): δ 4.62 (2H, s), 2.59 (1H, m), 1.20 (2H, m), 1.02 (2H, m). |
| 89% |
With ammonia; In tetrahydrofuran; at 0 - 20℃; for 17h; |
Ammonia gas was bubbled through THF (355 mL) at 0 C. for 20 min. Neat cyclopropylsulfonyl chloride (1a, 15 g, 0.11 mol) was dropwise added to the solution. The resulting solution was allowed to warm to room temperature and stirred for 17 h. The resulting suspension was filtered through a plug of silica gel, eluting with ethyl acetate. The filtrate was concentrated in vacuo to afford 11.5 g (89%) of cyclopropylsulfonamide 1b. 1H NMR (300 MHz, CD3OD): δ 2.63-2.53 (m, 1H), 1.09-0.95 (m, 4H). |
| 87% |
With ammonia; In tetrahydrofuran; at 0 - 20℃; |
[00279] Ammonia gas was bubbled through a gas dispersion tube into THF (40 mL) cooled to 0 0C for 5 minutes. To this solution at 0 0C was added cyclopropylsulfonylchloride (1 gram, 7.1 mmol). The reaction was stirred at room temperature overnight, then filtered through a plug of silica gel, followed by elution with EtOAc to yield 750 mg (6.19 mmol, EPO <DP n="294"/>87%) of cyclopropylsulfonamide. 1H-NMR (500 MHz, Methanol-d4): 4.79 (s, 2H), 2.59-2.54 (m, IH), 1.06-0.96 (m, 4H). |
| 74% |
With ammonia; In 1,4-dioxane; at 20℃; for 72h; |
Example 46 Compound of Formula IV, wherein Step 46a: Cyclopropylsulfonyl chloride (1.4 g, 10 mmol) was dissolved in 0.5 M ammonia in dioxane (50 ml, 25 mmol) at RT. The reaction was kept at RT for 3 days. The large amount of precipitation was filtered and discarded. The clear filtrate was evaporated in vacuo and the white residue was dried on vacuum for 24 hours to give the cyclopropylsulfonamide (0.88 g, 74%). 1H-NMR (500 MHz, CD3Cl): δ 4.62 (2H, s), 2.59 (1H, m), 1.20 (2H, m), 1.02 (2H, m). |
| 74% |
With ammonia; In 1,4-dioxane; at 20℃; for 72h; |
Cyclopropylsulfonyl chloride (1.4g, 10 mmol) was dissolved in 0.5 M ammonia in dioxane (50 ml, 25 mmol) at rt. The reaction was stirred at rt for 72 h. The precipitate was filtered and discarded. The clear filtrate was evaporated in vacuo and the white residue was dried on vacuum for 24 h to give cyclopropylsulfonamide (0.88 g, 74%).1H NMR (500 MHz, CD3Cl): δ 4.62 (2H, s), 2.59 (IH, m), 1.20 (2H, m), 1.02 (2H, m). |
| 74% |
With ammonia; In 1,4-dioxane; at 20℃; for 72h; |
Cyclopropylsulfonyl chloride (1.4g, 10 mmol) was dissolved in 0.5 M ammonia in dioxane (50 ml, 25 mmol) at rt. The reaction was stirred at rt for 72 h. The precipitate was filtered and discarded. The clear filtrate was evaporated in vacuo and the white residue was dried on vacuum for 24 h to give cyclopropylsulfonamide (0.88 g, 74%).1H NMR (500 MHz, CD3Cl): δ 4.62 (2H, s), 2.59 (IH, m), 1.20 (2H, m), 1.02 (2H, m). |
| 74% |
With ammonia; In 1,4-dioxane; at 20℃; for 72h; |
Cyclopropylsulfonyl chloride (1.4 g, 10 mmol) was dissolved in 0.5 M ammonia in dioxane (50 ml, 25 mmol) at RT. The reaction was kept at RT for 3 days. The large amount of precipitation was filtered and discarded. The clear filtrate was evaporated in vacuo and the white residue was dried on vacuum for 24 hours to give the cyclopropylsulfonamide (0.88 g, 74%). 1H-NMR (500 MHz, CD3Cl): δ 4.62 (2H, s), 2.59 (1H, m), 1.20 (2H, m), 1.02 (2H, m). |
| 74% |
With ammonia; In 1,4-dioxane; at 20℃; for 72h; |
Cyclopropylsulfonyl chloride (IAg, 10 mmol) was dissolved in 0.5 M ammonia in dioxane (50 ml, 25 mmol) at RT. The reaction was kept at RT for 3 days. The large amount of precipitation was filtered and discarded. The clear filtrate was evaporated in vacuo and the white residue was dried on vacuum for 24 hours to give the cyclopropylsulfonamide (0.88 g, 74%). 1H- NMR (500 MHz, CD3Cl): δ 4.62 (2H, s), 2.59 (IH, m), 1.20 (2H, m), 1.02 (2H, m). |
| 74% |
With ammonia; In 1,4-dioxane; at 20℃; for 72h; |
Step 3A: Cyclopropylsulfonyl chloride (1.4g, 10 mmol) was dissolved in 0.5 M ammonia in dioxane (50 ml, 25 mmol) at RT. The reaction was kept at RT for 3 days. The large amount of precipitation was filtered and discarded. The clear filtrate was <n="44"/>evaporated in vacuo and the white residue was dried on vacuum for 24 hours to give the cyclopropylsulfonamide (0.88 g, 74%). 1H-NMR (500 MHz, CD3Cl): δ 4.62 (2H, s), 2.59 (IH, m), 1.20 (2H, m), 1.02 (2H, m). |
| 52% |
With ammonium hydroxide; In methanol; at 20℃; for 16h; |
General procedure: Standard Procedure D for the Preparation of Sulfonamides (0054) A solution of sulfonyl chloride in methanol and ammonium hydroxide solution was stirred at 0 C. or room temperature. After the reaction was complete, methanol was removed under reduced pressure. The solution was extracted with ethyl acetate. The combined organic layers were dried over MgSO4(s), filtered, and concentrated to give the desired products without further purification. Step 1. Cyclopropanesulfonamide Following standard procedure D, cyclopropanesulfonyl chloride (0.400 mL, 3.95 mmol), methanol (3.0 mL), and ammonium hydroxide solution (15 mL) were used to carry out the reaction. After the reaction was stirred at room temperature for 16 h and work up, cyclopropanesulfonamide (0.249 g, 52%) was obtained as a white solid. 1H NMR (DMSO-d6, 300 MHz) δ 6.78 (br s, 2H), 2.50-2.46 (m, 1H), 0.89-0.86 (m, 4H). |
| 3.45 g (100%) |
In tetrahydrofuran; ammonia; |
Preparation of cyclopropyl sulfonamide To a solution of 100 mL of THF cooled to 0 C. was bubbled in gaseous ammonia until saturation was reached. To this solution was added a solution of 5 g (28.45 mmol) of cyclopropylsulfonyl chloride (purchased from Array Biopharma) in 50 mL of THF, the solution warmed to rt overnite and stirred one additional day. The mixture was concentrated until 1-2 mL of solvent remained, applied onto 30 g plug of SiO2 (eluted with 30% to 60% EtOAc/Hexanes) to afford 3.45 g (100%) of cyclopropyl sulfonamide as a white solid. 1H NMR (Methanol-d4) δ 0.94-1.07 (m, 4H), 2.52-2.60 (m, 1H); 13C NMR (methanol-d4) δ 5.92, 33.01. |
| 1.70 g (99%) |
In tetrahydrofuran; ammonia; |
Preparation of Cyclopropyl Sulfonamide Moiety for Use in Step 2e: To a solution of 20 mL THF cooled to 0 C. was bubbled in gaseous ammonia until saturation was reached. To this solution was added 2 g (5.69 mmol) of cyclopropylsulfonyl chloride (purchased from Array Biopharma). The solution was warmed to room temperature over two hours and the crude solution was filtered through a plug of silica gel eluding the product with ethyl acetate. The fractions were concentrated in vacuo to yield 1.70 g (99%) of cyclopropyl sulfonamide as a white solid: 1H NMR (d4-MeOH, 500 MHz) δ 0.94-1.07 (m, 4H), 2.52-2.60 (m, 1H). |
| 3.45 g (100%) |
In tetrahydrofuran; ammonia; |
Method 2 To a solution of 100 mL of THF cooled to 0 C. was bubbled in gaseous ammonia until saturation was reached. To this solution was added a solution of 5 g (28.45 mmol) of cyclopropylsulfonyl chloride (purchased from Array Biopharma) in 50 mL of THF. The solution was warmed to room temperature overnight and stirred one additional day. The mixture was concentrated until 1-2 mL of solvent remained and poured onto a 30 g plug of SiO2 (eluted with 30% to 60% ethyl acetate/hexanes) to provide 3.45 g (100%) of cyclopropylsulfonamide as a white solid. 1H NMR (methanol-d4) δ 0.94-1.07 (m, 4H), 2.52-2.60 (m, 1H); 13C NMR (methanol-d4) δ 5.92, 33.01. |
| 3.45 g (100%) |
In tetrahydrofuran; ammonia; |
Method 2 To a solution of 100 mL of THF cooled to 0 C. was bubbled in gaseous ammonia until saturation was reached. To this solution was added a solution of 5g (28.45 mmol) of cyclopropylsulfonyl chloride (purchased from Array Biopharma) in 50 mL of THF. The solution was warmed to room temperature overnight and stirred one additional day. The mixture was concentrated until 1-2 mL of solvent remained and poured onto a 30 g plug of SiO2 (eluted with 30% to 60% ethyl acetate/hexanes) to provide 3.45 g (100%) of cyclopropylsulfonamide as a white solid. 1H NMR (methanol-d4) δ 0.94-1.07 (m, 4H), 2.52-2.60 (m, 1H); 13C NMR (methanol-d4) δ 5.92, 33.01. |
|
With ammonia; In dichloromethane; at 0 - 20℃; |
To a solution of cyclopropylmagnesium bromide (0 5 M 20 mL, 10 0 mmol) in anhydrous THF (10 mL) at about -100C is added a solution of SO; in THF (~ 16 wt%, 4 8 mL, 12 mmo.) slowiy over 10 mm at -10 to -5C Tne reaction mixture is warmed to ambient temperature over 0 5h, and then NCS (2 O g 15 mmo.) is added at about -5 to 0C The reaction mixture is warmed up to ambient temperature and diluted with 50 mL oi methyl terl-buty. ether To tne reaction mixture ι≤ added 50 mL water and the mixture is stirred for 5 mm The organic layer is washed with 50 mL of bnne The organic layer is concentrated and the resultant cyclopropylsuifony. chloride is dissolved in CH2Ci2 (total volume was about 50 mL) and ammonia gas is bubbled in at about O'C for about 5 mm and the mixture is slowly warmed up to ambient temperature and stored at that temperature for 2 h The mixture is filtered through Cehte to remove the solid (NH4C.), and the filtrate is concentrated to obtain a crude cyclopropyisulfonamide solid (~ 1 2g) Re-cryslailization of the crude product from EtOAc/hexane produces cyclopropyisuifonarnide 42 in 80% overall yield. |
| 7.03 g |
With ammonia; In tetrahydrofuran; for 16h; |
Cyclopropanesulfonamide A mixture of 50 ml of 25% aqueous ammonia solution and 50 ml of THF was initially charged, and then 10.00 g of cyclopropanesulfonyl chloride in 10 ml of THY were slowly added dropwise and the mixture was stirred for 16 hours. After concentration by rotary evaporation and coevaporation with toluene, the residue was extracted by stirring with 100 ml of ethyl acetate and the solids were filtered off. The organic phase was dried over Na2SO4 and concentrated by rotary evaporation. This gave the product (7.03 g) with a molecular weight of 121.2 g/mol (C3H7NO2S); MS (ESI): m/e=122 (M+H+). |
| 7.03 g |
With ammonium hydroxide; In tetrahydrofuran; water; for 16h; |
Cyclopropanesulfonamide A mixture of 50 ml of 25% aqueous ammonia solution and 50 ml of THF was initially charged, and then 10.00 g of cyclopropanesulfonyl chloride in 10 ml of THF were slowly added dropwise and the mixture was stirred for 16 hours. After concentration by rotary evaporation and coevaporation with toluene, the residue was extracted by stirring with 100 ml of ethyl acetate and the solids were filtered off. The organic phase was dried over Na2SO4 and concentrated by rotary evaporation. This gave the product (7.03 g) with a molecular weight of 121.2 g/mol (C3H7NO2S); MS (ESI): m/e=122 (M+H+). |
| 7.03 g |
In tetrahydrofuran; water; for 16h; |
A mixture of 50 ml of 25% aqueous ammonia solution and 50 ml of THF was initially charged, and then 10.00 g of cyclopropanesulfonyl chloride in 10 ml of THF were slowly added dropwise and the mixture was stirred for 16 hours. After concentration by rotary evaporation and coevaporation with toluene, the residue was extracted by stirring with 100 ml of ethyl acetate and the solids were filtered off. The organic phase was dried over Na2SO4 and concentrated by rotary evaporation. This gave the product (7.03 g) with a molecular weight of 121.2 g/mol (C3H7NO2S); MS (ESI): m/e=122 (M+H+). |
|
With ammonia; In tetrahydrofuran; at 0 - 20℃; |
[0314] Ammonia gas was bubbled through a gas dispersion tube into THF (40 mL) cooled to 0 0C for 5 minutes. To this solution at 00C was added cyclopropylsulfonylchloride (1 gram, 7.1 mmol). The reaction was stirred at room temperature overnight, then filtered through a plug of silica gel, followed by elution with EtOAc to yield 750 mg (6.19 mmol) of cyclopropylsulfonamide. 1H-NMR (500 MHz, Methanol-d4): 4.79 (s, 2H), 2.59-2.54 (m, IH), 1.06-0.96 (m, 4H). |






 Chemistry
Chemistry
 Pharmaceutical Intermediates
Pharmaceutical Intermediates
 Inhibitors/Agonists
Inhibitors/Agonists
 Material Science
Material Science






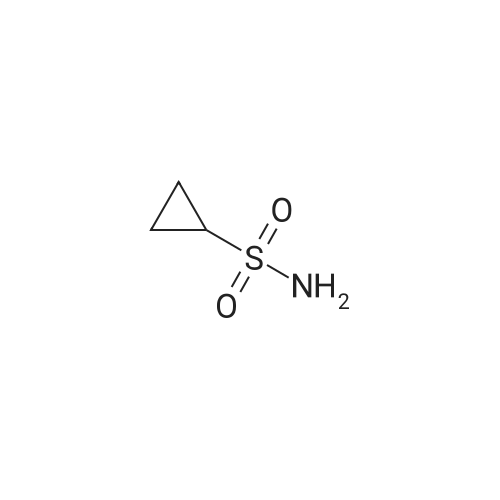

 For Research Only
For Research Only
 120K+ Compounds
120K+ Compounds
 Competitive Price
Competitive Price
 1-2 Day Shipping
1-2 Day Shipping