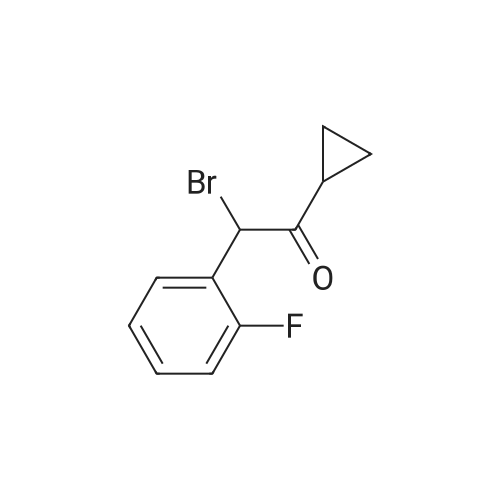
| 90.4% |
With bromine; In tetrahydrofuran; at 20℃; for 10h; |
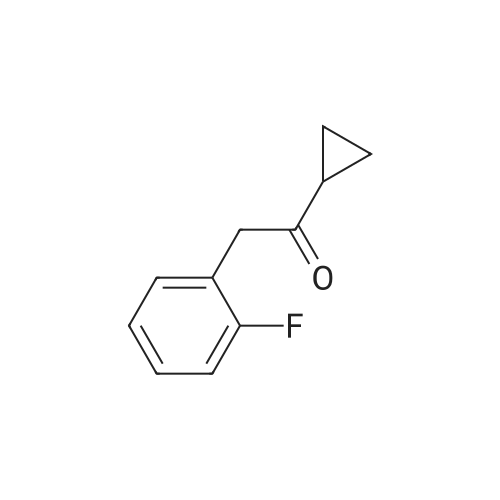
Prasugrel intermediate 1-cyclopropyl -2 bromo-2 - (2-fluoro phenyl) - 2-ethanone synthesis of The 1-cyclopropyl-2 - (2-fluoro phenyl) - 2-ethyl ketone 29.1g (10mmol) with bromine (20mmol) in the tetrahydrofuran 20 C reaction 10 hours, after the reaction, cooling to room temperature, into ice water, dichloromethane extraction, saturated sodium bisulfite washing, organic phase is concentrated, anhydrous methanol is recrystallized to get pulls Grey intermediate 1-cyclopropyl-2-bromo-2 - (2-fluoro phenyl) - 2-ethyl ketone 2.3g, the yield is 90.4%, purity of 99.86%. |
| 83.2% |
With N-Bromosuccinimide; 2,2'-azobis(isobutyronitrile); In chloroform; at 20℃; for 8h; |
8.9 g of cyclopropyl-2-(2-fluorophenyl)ethanone was dissolved in 80 ml of chloroform.Add three reaction flasks, add 8% of azobisisobutyronitrile, and add 9.6g of NBS in batches under stirring.After the addition was completed at 20C for 8 hours, the other steps were the same as in Example 2 to obtain 10.7 g of compound (V) in a yield of 83.2% and HPLC: >96.4%. |
| 78% |
With N-Bromosuccinimide; toluene-4-sulfonic acid; at 80℃;Product distribution / selectivity; |
Example 32-bromo-l-cycIopropyI-2-(2-fluorophenyI)-ethanone (compound of the Formula (III) )4.0 g of l-cyclopropyl-2-(2-fluorophenyl)-ethanone having 98.0% content according to gas chromatographic analysis is introduced into a 100-ml round-bottom flask and 0.4 g of p-toluenesulfonic acid monohydrate are added. The mixture is heated to 80 C and at the same temperature, 3.8 g of N-bromosuccinimide are added. After the reaction is complete, the mixture is cooled to a temperature between 20 and 25 C. 18 g of 5 % by weight sodium hydroxide solution and 30 ml of methyl-t-butylether are added. The mixture is stirred for 10 minutes and subsequently the layers are separated. The organic layer is washed twice with 20 ml of water each and the organic layer is evaporated.Yield of the raw product: 5.7 g (pure: 4.5 g, 78%), pale yellow liquid. Assay (measured by gas chromatography) 79.3% |
| 73.8% |
With hydrogen bromide; dihydrogen peroxide; In ethanol; water; for 0.5h;Heating / reflux;Product distribution / selectivity; |
In a 250 ml round-bottomed flask a solution of cyclopropyl-2-fluorbenzyl ketone (8.91 g, 50 mmol), ethanol (50 ml), aqueous hydrogen peroxide solution (30 w%, 30 ml, 0.29 mol) and aqueous hydrogen bromide solution (48 w%, 22.6 ml, 0.20 mol) are added. The reaction mixture is boiled for half an hour and the colourless solution is evaporated. To the residue water (50 ml) and ethyl acetate (50 ml) are added at 25 C, the phases are separated and the organic phase is dried and evaporated.The obtained product: 9.4 g light yellow oil Yield: 73.8 %Content (measured by GC/MS): <n="17"/>83.4 % title product, which is contaminated with 15.2 % starting compound and 0.5 % dibromo derivative. |
| 67.1% |
With hydrogen bromide; dihydrogen peroxide;benzyltriethylammonium bromide; In water; at 85℃; for 2h;Product distribution / selectivity; |
In a 250 ml round-bottomed flask a solution of cyclopropyl-2-fluorbenzyl ketone (8.91 g, 50 mmol), 7V-benzyl-triethyl-ammonium bromide (2.0 g), aqueous hydrogen peroxide solution (30 w%, 40 ml, 0.39 mol) and aqueous hydrogen bromide solution (48 w%, 28.3 ml, 0.25 mol) are added. The reaction mixture is stirred intensively for two hours at 85 0C and the product is extracted twice with ethyl acetate (2x50 ml) and the united phases of ethyl acetate are dried and evaporated.The obtained product: 10.2 g light yellow oilYield: 67.1 %Content (measured by GC/MS):83.4 % title product, which is contaminated with 7.1 % starting compound and 7.5 % dibromo derivative. |
|
With N-Bromosuccinimide; dibenzoyl peroxide; In tetrachloromethane; toluene; |
(b) 5-(alpha-Cyclopropylcarbonyl-2-fluorobenzyl)-2-oxo-2,4,5,6,7,7a-hexahydrothieno[3,2-c]pyridine To a solution of cyclopropyl 2-fluorobenzyl ketone (8.7 g) obtained in Reference example 1(a) in carbon tetrachloride (80 ml) was added N-bromosuccinimide (9.6 g) and benzoyl peroxide (0.5 g), then the mixture was heated under reflux for 6 hours. After the reaction, toluene was added to the reaction mixture and the resulting solid was filtered off. The filtrate was concentrated under reduced pressure. The residue was purified by chromatography on a silica gel column using toluene as the eluant to afford alpha-cyclopropylcarbonyl-2-fluorobenzyl bromide (8.5 g) as a yellow oil. |
|
With N-Bromosuccinimide; dibenzoyl peroxide; In tetrachloromethane; for 6h;Heating / reflux; |
To a solution of cyclopropyl 2-fluorobenzyl ketone (8.7 g) obtained in part (a) in carbon tetrachloride (80 ml) was added N-bromosuccinimide (9.6 g) and benzoyl peroxide (0.5 g), then the mixture was heated under reflux for 6 hours. After the reaction, toluene was added to the reaction mixture and the resulting solid was filtered off. The filtrate was concentrated under reduced pressure. The residue was purified by chromatography on a silica gel column using toluene as the eluant to afford alpha-cyclopropylcarbonyl-2-fluorobenzyl bromide (8.5 g) as a yellow oil. To a solution of alpha-cyclopropylcarbonyl-2-fluorobenzyl bromide (6.0 g) obtained above in dimethylformamide (20 mL) was added 2-oxo-2,4,5,6,7,7a-hexahydrothieno[3,2-c]pyridine hydrochloride (4.8 g), which was prepared according to the method described in EP 192535 (Japanese Patent Application Publication No. Sho 61-246186) and potassium bicarbonate (7.0 g). After stirring the mixture at room temperature for 2 hours the reaction mixture was partitioned between ethyl acetate and water. The ethyl acetate layer washed with saturated aqueous sodium chloride solution, then dried over anhydrous magnesium sulfate, and evaporated under reduced pressure. After purification of the residue by chromatography on a silica gel column using toluene/ethyl acetate=3/1 as the eluant, the product was crystallized from diisopropyl ether to afford the desired product (2.6 g, yield 35%) as pale brown crystals. 1H NMR (CDCl3) delta ppm: 0.75-0.96 (2H, m), 0.99-1.14 (2H, m), 1.83-2.01 (1H, m), 2.02-2.17 (1H, m), 2.25-2.45 and 2.47-2.62 (total 2H, each m), 2.85 and 3.10 (total 2H, each d, J=12.0 Hz), 3.88-4.01 and 4.03-4.16 (total 2H, each m), 4.85 and 4.89 (total 1H, each s), 6.03 and 6.06 (total 1H, each s), 7.10-7.45 (4H, m). Mass (Cl, m/z):332 (M ++1), 262; Anal Calcd. for C18H18FNO2S: C, 65.23; H, 5.48; N, 4.23. Found: C, 65.09; H, 5.55; N, 4.20. |
|
With N-Bromosuccinimide; dibenzoyl peroxide; In chloroform; for 6h;Heating / reflux; |
Example-25: Preparation of 2-fluoro- alpha-cyclopropyl carbonyl benzyl bromide; A mixture of l-cyclopropyl-2-(2-fluorophenyl)ethanone (40 grams), chloroform (400 ml), N-bromo succinamide (50 grams) and benzoylperoxide (0.6 gram) was heated to reflux temperature and stirred for 6 hours. The reaction mixture was cooled to 25-300C and then filtered and the solid washed with ,. chloroform. The filtrate washed with water and then distilled off the filtrate under reduced pressure. The residue was cooled to 25-300C and extracted the product with cyclohexane. The extracted layer was distilled under reduced pressure to get the title compound. Yield: 45 grams; |
|
With hydrogen bromide; dihydrogen peroxide; In 1,4-dioxane; water; at 20 - 85℃; for 2 - 120h;Product distribution / selectivity; |
hi a 1000 ml round-bottomed flask a solution of cyclopropyl-2-fluorobenzyl ketone (44.6 g, 0.25 mol), dioxane (500 ml), aqueous hydrogen peroxide solution (30 w/w%, 125 ml, 1.22 mol) and aqueous hydrogen bromide solution (48 w/w%, 71.3 ml, 0.63 mol) are added. The reaction mixture is warming to 50 0C and it is stirred for two hours at a temperature of 80-85 C. To the colourless solution sodium sulfate (20 g) is added at 25 C, it is stirred until dissolution, extracted and the organic layer is washed with aqueous sodium hydrogen carbonate (5 w/w %, 150 ml), separated and the organic layer is dried over magnesium sulfate and evaporated. <n="14"/>The obtained product: 59.2 g light yellow oilYield: 82.9 %Content (measured by GC): 87.1 %. According to the results of the GC examination, it contains 7.5 % of the starting compound, 2.5 % of the monobromo impurity and 1.3 % of the dibromo derivatives. The product is purified by vacuum distillation.Boiling point: 90 0C / 0.3 HgmmThe obtained product after distillation: 49.1 g colourless oil Content of the obtained title product (measured by GC) after distillation: 97.5 %IR (film): 3405, 3011, 1713, 1614, 1587, 1491, 1458, 1380, 1235, 1196, 1068,1023.1H-NMR (CDCl3, 500 MHz): 7.49 (t, IH), 7.33 (m, IH), 7.18 (t, IH), 7.08 (t, IH),5.96 (s, IH), 2.14 (m, IH), 1.17 (m, IH), 1.11 (m, IH), 1.02 (m, IH), 0.94 (m,IH).13C-NMR (CDCl3, 125 MHz): 200.5, 159.7 (d, J = 249.0 Hz), 131.1 (d, J = 2.4Hz), 130.8 (d, J = 8.3 Hz), 124.7 (d, J = 3.9 Hz), 123.4 (d, J = 13.2 Hz), 115.6 (d,J = 22.0 Hz), 48.3 (d, J = 2.9 Hz), 18.7, 12.7, 12.6; The preparation process is carried out according to example 1. with the difference that aqueous hydrogen bromide solution (48 w%, 71.3 ml, 0.63 mol) is added dropwise to the starting reaction mixture at 25 0C under cooling and under intensive stirring. The obtained mixture is stirred for 5 days at room temperature and the product is prepared according to example 1. <n="18"/>The obtained product: 59.2 g light yellow oilYield: 82.9 %Content (measured by GC): 90.0 %. According to the measurement by GC the crude product contains 7.2 % starting product and only 1.3 % dibromo derivative, which is less than the obtained bromo derivative in example 1. The product, if it is necessary, can be purified by distillation.Boiling point: 90 0C / 0.3 HgmmThe obtained product after distillation: 49.1 g colourless oilContent of (GC) after distillation: 97.5 % |
|
With N-Bromosuccinimide; In dichloromethane; for 10h;Heating / reflux;Product distribution / selectivity; |
2. Preparation of compound of formula (TV) with iV-bromo- succinimide:The process described in United States Patent Application No. 2003/134872 was reproduced, wherein the bromination was carried out with N- bromo-succinimide in carbon tetrachloride and the crude product was measured by GC/MS method. According to the GC/MS measurement the reaction mixture contained68.5 % compound of formula (IV),5.8 % monobromo compound, derived from the opening of the cyclopropane ring, 5.5 % dibromo compound, derived from the opening of the cyclopropane ring,16.0 % unreacted starting compound.The GC chromatogram of Figure I demonstrates the significant amount of impurities in the crude product. <n="10"/>The same reaction was carried out in dichloromethane, instead of carbon tetrachloride, and the reaction mixture was refluxed for 10 hours. The obtained amount of impurities was almost the same. |
|
With N-Bromosuccinimide; In tetrachloromethane;Product distribution / selectivity; |
2. Preparation of compound of formula (TV) with iV-bromo- succinimide:The process described in United States Patent Application No. 2003/134872 was reproduced, wherein the bromination was carried out with N- bromo-succinimide in carbon tetrachloride and the crude product was measured by GC/MS method. According to the GC/MS measurement the reaction mixture contained68.5 % compound of formula (IV),5.8 % monobromo compound, derived from the opening of the cyclopropane ring, 5.5 % dibromo compound, derived from the opening of the cyclopropane ring,16.0 % unreacted starting compound.The GC chromatogram of Figure I demonstrates the significant amount of impurities in the crude product. <n="10"/>The same reaction was carried out in dichloromethane, instead of carbon tetrachloride, and the reaction mixture was refluxed for 10 hours. The obtained amount of impurities was almost the same. |
|
With pyridine; bromine; In dichloromethane; at 20℃; for 12h;Product distribution / selectivity; |
1. Preparation of compound of formula (TV) with bromine:A compound of formula (II) was reacted with an equimolar amount of bromine in dichloromethane, at room temperature for 12 hours, until the colour of the bromine disappeared. According to the GC/MS measurement the reaction mixture contained15 % compound of formula (IV),35 % monobromo compound, derived from the opening of the cyclopropane ring,17 % dibromo compound, derived from the opening of the cyclopropane ring, and <n="9"/>19 % unreacted starting compound.When pyridine was added to the reaction mixture, the obtained amount of compound of formula (IV) increased to 30 %, but the ratio of the compounds in the mixture was similar to the above described composition.The same reaction was carried out in acetic acid. According to the GC/MS measurement, the reaction mixture contained only 3.5 % compound of formula (IV). The content of the mixture was the following:3.5 % compound of formula (IV),15 % monobromo compound, derived from the opening of the cyclopropane ring,47 % dibromo compound, derived from the opening of the cyclopropane ring, and31 % unreacted starting compound.The conclusion of the above experiments is that compound of formula (IV) can not be prepared with bromine in a good yield. |
|
With bromine; In dichloromethane; at 20℃; for 12h;Product distribution / selectivity; |
1. Preparation of compound of formula (TV) with bromine:A compound of formula (II) was reacted with an equimolar amount of bromine in dichloromethane, at room temperature for 12 hours, until the colour of the bromine disappeared. According to the GC/MS measurement the reaction mixture contained15 % compound of formula (IV),35 % monobromo compound, derived from the opening of the cyclopropane ring,17 % dibromo compound, derived from the opening of the cyclopropane ring, and <n="9"/>19 % unreacted starting compound.When pyridine was added to the reaction mixture, the obtained amount of compound of formula (IV) increased to 30 %, but the ratio of the compounds in the mixture was similar to the above described composition.The same reaction was carried out in acetic acid. According to the GC/MS measurement, the reaction mixture contained only 3.5 % compound of formula (IV). The content of the mixture was the following:3.5 % compound of formula (IV),15 % monobromo compound, derived from the opening of the cyclopropane ring,47 % dibromo compound, derived from the opening of the cyclopropane ring, and31 % unreacted starting compound.The conclusion of the above experiments is that compound of formula (IV) can not be prepared with bromine in a good yield. |
|
With bromine; In acetic acid;Product distribution / selectivity; |
1. Preparation of compound of formula (TV) with bromine:A compound of formula (II) was reacted with an equimolar amount of bromine in dichloromethane, at room temperature for 12 hours, until the colour of the bromine disappeared. According to the GC/MS measurement the reaction mixture contained15 % compound of formula (IV),35 % monobromo compound, derived from the opening of the cyclopropane ring,17 % dibromo compound, derived from the opening of the cyclopropane ring, and <n="9"/>19 % unreacted starting compound.When pyridine was added to the reaction mixture, the obtained amount of compound of formula (IV) increased to 30 %, but the ratio of the compounds in the mixture was similar to the above described composition.The same reaction was carried out in acetic acid. According to the GC/MS measurement, the reaction mixture contained only 3.5 % compound of formula (IV). The content of the mixture was the following:3.5 % compound of formula (IV),15 % monobromo compound, derived from the opening of the cyclopropane ring,47 % dibromo compound, derived from the opening of the cyclopropane ring, and31 % unreacted starting compound.The conclusion of the above experiments is that compound of formula (IV) can not be prepared with bromine in a good yield. |
|
With hydrogen bromide; dihydrogen peroxide; In water; acetic acid; at 95℃; for 1h;Product distribution / selectivity; |
In a 250 ml round-bottomed flask a solution of cyclopropyl-2-fluorbenzyl ketone (8.91 g, 50 mmol), acetic acid (50 ml), aqueous hydrogen peroxide solution (30 w%, 15 ml, 0.14 mol) and solution of hydrogen bromide in acetic acid (33 w%, <n="16"/>14.7 ml, 0.13 mol) are added. The reaction mixture is stirred for one hour at a temperature of 95 C. The colourless solution is diluted with water (150 ml), extracted with toluene (100 ml) and the organic layer is separated, dried and evaporated in vacuo.The obtained product: 11.4 g light yellow oilYield: 75.4 %Content (GC/MS): 85.0 %, it is contaminated with 5.2 % starting compound and 7.5 % dibromo derivative. The product is purified by vacuum distillation.Boiling point: 95 C / 0.4 HgmmThe obtained product after distillation: 8.3 g colourless oil Content of the obtained title product (measured by GC) after distillation: 98.5 % |
|
With N-Bromosuccinimide; 2,2'-azobis(isobutyronitrile); toluene-4-sulfonic acid; In tetrachloromethane; at 75℃; for 3h; |
EXAMPLE 5: PREPARATION OF 2-FLUORO-alpha-CYCLOPROPYL CARBONYL BROMIDE.Cyclopropyl-2-fluorobenzyl ketone (50 g), carbon tetrachloride (1000 mL), N-bromo succinamide (60 g), AIBN (3 g) and PTSA (1.5 g) are charged into a round bottom flask with stirring. The reaction mixture is heated to reflux (75 0C) and stirred for 3 hours. The reaction mixture is cooled to 15 0C and then the suspension is filtered. The filtrate is washed with 5% sodium bisulfate solution (2chi250 mL) and then the obtained organic layer is dried over sodium sulfate. The resultant organic layer is concentrated completely under vacuum at 50 0C to provide crude product. Hexane (300 mL) is charged to the obtained crude mass and stirred for 10 minutes followed by decantation of n-hexane. The obtained decanted n-hexane layer is distilled completely at 36 0C to afford 55.0 g of the title compound.Purity: 73.6% by HPLC. |
|
With N-Bromosuccinimide; 2,2'-azobis(isobutyronitrile); toluene-4-sulfonic acid; In chloroform; for 4h;Reflux; |
Example-18: Preparation of 2-bromo-l-cyclopropyl-2-(2-fluorophenyl) ethanone compound of forniula-3:A mixture of l-cyclopropyl-2-(2-fluorophenyl) ethanone (40 grams), chloroform (400 ml), N-bromo succinamide (50 grams), azobisisobutyronitrile (2.4 grams) and p-toluenesulfonic acid (1.2 grams) was stirred for 4 hrs at reflux temperature. The reaction mixture was cooled to 0-5C and stirred for 45 minutes and the precipitated solid was filtered. The filtrate was distilled off completely under reduced pressure. Cyclohexane (100 ml) was added to the obtained residue and distilled off the solvent under reduced pressure to get the title compound.Yield: 55 grams |
|
With hydrogen bromide; dihydrogen peroxide; In water; at 25 - 40℃; |
Example-7: Preparation of a-cyclopropylcarbonyl-2-fluorobenzyI bromide compound of formula-4: 142 ml of aqueous HBr was added to the solution of water (500 ml) and l-cyclopropyl-2-(2-fluorophenyl)ethanone (100 grams) at 25-30C and stirred for 15 minutes at the same temperature. 30% hydrogen peroxide solution (127 ml) was added to the reaction mixture at 25-30C. The reaction mixture was stirred for 2 hours at 25-30C and raised the temperature of the reaction mixture to 35-40C. Stirred the reaction mixture for 4 hours at the same temperature. After completion of the reaction mixture, the reaction mixture was cooled to 10-20C. The reaction mixture was poured into pre-cooled water at below 25C and cyclohexane was added to the reaction mixture. Both the organic and aqueous layers were separated. The organic layer was washed with 5% sodium thiosulphite solution followed by 5% potassium iodide solution and water. The solvent was distilled off under reduced pressure completely from the organic layer and then get the title compound.Yield: 140 grams. |
|
With N-Bromosuccinimide; dibenzoyl peroxide; In ethyl acetate; at 25 - 60℃;Product distribution / selectivity; |
Method AIn a 2 ltr 4-necked flask equipped with a thermometer and mechanical stirrer, N-Bromo succinamide(125 g, 0.55 moles) and benzoyl peroxide (1.8 g, 0.007 moles) was added to a solution of cyclopropyl 2-fluorobenzyl ketone (100 g, 0.56 moles) in Ethyl acetate at 25 C (+/-5 C). Stir the reaction mass for 30 to 40 hrs at 60 C (+/-2 C). Cool the reaction mass and filter through hyflo. Recover the solvent under vacuum and dissolved into petroleum ether, hexane, heptanes or mixture thereof. Cool to 5 C (+/-2 C) and separate the brown colour lower layer, if any. Recover the solvent under vacuum to obtain pure 2-Fluoro-a-cyclopropylcarbonylbenzyl bromide (140 g, HPLC purity 95%) |
|
With N-Bromosuccinimide;dibenzoyl peroxide; In 1,2-dichloro-ethane; for 7h;Reflux; |
Cyclopropyl 2-fluorobenzyl ketone (50 gm), ethylene dichloride (500 ml), n- bromosuccinate (80 gm) and benzoyl peroxide (1 gm) were added at room temperature.The contents were heated to reflux and maintained for 7 hours at reflux. The reaction mass was cooled to 0C and filtered. The solvent was distilled off under vacuum at below 45C to obtain residual mass. The residual mass was dissolved in carbon tetrachloride (200 ml) at room temperature and stirred for 30 minutes. The separated solid was filtered and concentrated to obtain 65 gm of cyclopropyl-2-fluorobenzyl carbonyl bromide |
|
With 1,3-dibromo-5,5-dimethylimidazolidine-2,4-dione; 2,2'-azobis(isobutyronitrile); acetic acid; at 60 - 85℃; for 2.41667h; |
13.1g o-fluorobenzyl cyclopropyl ketone and 40 ml acetic acid were added to a 100ml four-neck flask equipped with a mechanical stirring device, a thermometer, a reflux condenser and a constant-pressure dropping funnel. 12.1 g 1,3-dibromo-5,5-dimethylhydantoin and 0.66g azobisisobutyronitrile were added in dropwise to the above reaction system at 60C?85C over 2 hours. After the completion of addition, the mixture was kept at this temperature and stirred for 25 minutes. Next, the mixture was cooled and distilled to remove most of acetic acid. 40ml ethyl acetate and 40ml water were added to the concentrated solution, and the system was allowed to seperation. The organic layer was washed with 20ml saturated Na2SO3, 20ml saturated NaHCO3 and 20ml saturated brine successively, and then dried over anhydrous magnesium sulfate. The filtrate was distillated under reduced pressure and concentrated to give 21.1g brown oil. The yield was 83.5% and the purity was 74.8%. |
|
With 1,3-dibromo-5,5-dimethylimidazolidine-2,4-dione; acetic acid;2,2'-azobis(isobutyronitrile); at 60 - 85℃; for 2.41667h; |
13.1 g o-fluorobenzyl cyclopropyl ketone and 40 ml acetic acid were added to a 100 ml four-neck flask equipped with a mechanical stirring device, a thermometer, a reflux condenser and a constant-pressure dropping funnel. 12.1 g 1,3-dibromo-5,5-dimethylhydantoin and 0.66 g azobisisobutyronitrile were added in dropwise to the above reaction system at 60 C.-85 C. over 2 hours. After the completion of addition, the mixture was kept at this temperature and stirred for 25 minutes. Next, the mixture was cooled and distilled to remove most of acetic acid. 40 ml ethyl acetate and 40 ml water were added to the concentrated solution, and the system was allowed to seperation. The organic layer was washed with 20 ml saturated Na2SO3, 20 ml saturated NaHCO3 and 20 ml saturated brine successively, and then dried over anhydrous magnesium sulfate. The filtrate was distillated under reduced pressure and concentrated to give 21.1 g brown oil. The yield was 83.5% and the purity was 74.8%. |
|
With N-Bromosuccinimide; 2,2'-azobis(isobutyronitrile);toluene-4-sulfonic acid; In chloroform; at 0 - 5℃; for 4.75h;Reflux; |
Example 18Preparation of 2-bromo-1-cyclopropyl-2-(2-fluorophenyl)ethanone Compound of Formula-3 A mixture of 1-cyclopropyl-2-(2-fluorophenyl) ethanone (40 grams), chloroform (400 ml), N-bromo succinamide (50 grams), azobisisobutyronitrile (2.4 grams) and p-toluenesulfonic acid (1.2 grams) was stirred for 4 hrs at reflux temperature. The reaction mixture was cooled to 0-5 C. and stirred for 45 minutes and the precipitated solid was filtered. The filtrate was distilled off completely under reduced pressure. Cyclohexane (100 ml) was added to the obtained residue and distilled off the solvent under reduced pressure to get the title compound.Yield: 55 grams |
|
With N-Bromosuccinimide; 2,2'-azobis(isobutyronitrile);toluene-4-sulfonic acid; In dichloromethane; at 0 - 5℃; for 4.75h;Reflux; |
Example 32Preparation of 2-bromo-1-cyclopropyl-2-(2-fluorophenyl) ethanone Compound of Formula-3 A mixture of 1-cyclopropyl-2-(2-fluorophenyl) ethanone (40 grams), methylene chloride (400 ml), N-bromo succinamide (50 grams), azobisisobutyronitrile (2.4 grams) and p-toluenesulfonic acid (1.2 grams) was stirred for 4 hrs at reflux temperature. The reaction mixture was cooled to 0-5 C. and stirred for 45 minutes and the precipitated solid was filtered. The filtrate was distilled off completely under reduced pressure. Cyclohexane (100 ml) was added to the obtained residue and distilled off the solvent under reduced pressure to get the title compound.Yield: 55 grams |
|
With N-Bromosuccinimide; toluene-4-sulfonic acid; In acetonitrile; at 40℃; for 24h; |
Into a 100 ml round-bottomed flask 4.0 g of 98 % (according to GC analysis) of 1-cyclopropyl- 2-(2-fluorophenyl)-ethanone, and thereafter 0.4 g of p-toluenesulfonic acid monohydrate and 12 ml of acetonitrile are added. The mixture is warmed to 40C and at this temperature a solution of 4.0 g N-bromo-succinimide and 32 ml of acetonitrile is added. The reaction mixture is stirred at 40C for 24 hours, then cooled to 20-25C, whereupon 18 g of a 5 vol.% sodium hydroxide solution and 30 ml of tert. butyl -methyl-ether are added. The reaction mixture is stirred for 10 minutes and the phases are separated. The organic layer is washed twice with 20 ml of water each. The organic layer is evaporated. Thus 5,5 g of the desired product are obtained /main component 5.0 g ( 87 %). Pale yellow liquid. GC content 91.0 %. |
|
With bromine; In methanol;Large scale; |
To 230 1 methanol 17.82 kg l-cyclopropyl-2-(2-fluorophenyl)ethanone is measured. To the reaction mixture with continuous stirring in a period of 2-2.5 hour, 15.98 kg bromine is added gradually, the reaction mixture is cooled to 0-5C temperature, then 230 1 dichloromethane and 8,4 kg sodium-hydrogen-carbonate, and 230 1 water are added. The phases are separated and the aqueous phase is washed with 100 1 dichloromethane. The combined organic phase is washed with 1001 water, dried and evaporated at reduced pressure. (0076) 26.11 kg title compound is obtained (corrected with the content: 24.62 kg, yield: 95.8%)), the concentration of l,5-dibromo-l-(2-fluoropheny)pentane-2-one of Formula (TX) is not more than 0.05% (with capillary gas-chromatography). |






 Chemistry
Chemistry
 Pharmaceutical Intermediates
Pharmaceutical Intermediates
 Inhibitors/Agonists
Inhibitors/Agonists
 Material Science
Material Science







 For Research Only
For Research Only
 120K+ Compounds
120K+ Compounds
 Competitive Price
Competitive Price
 1-2 Day Shipping
1-2 Day Shipping