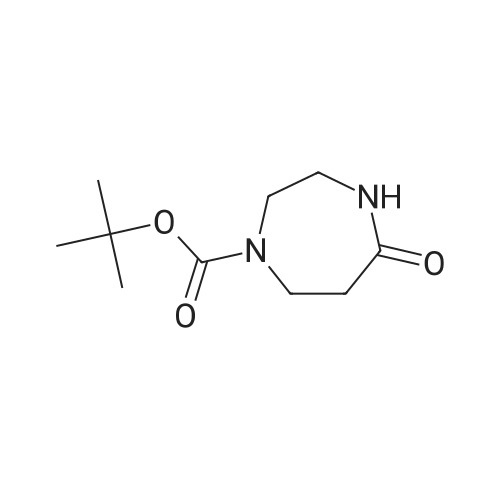
| 65% |
With hydroxylamine hydrochloride; In pyridine; at 65℃; for 1h; |
To a solution of N-t-butoxycarbonyl-4-piperidone 1 (15 g, 75.3 mmol) in pyridine (50 mL) was added hydroxylamine ? HCl (5.23 g, 75.3 mmol). The mixture was heated in an oil bath at 65 C for 1 h. After cooling, pyridine was removed under reduced pressure and the residue was dried under high vacuum overnight to give a solid. To this solid was added water (100 mL) and the mixture was sonicated. The precipitate was filtered and washed with water then dried under high vacuum to give the oxime derivative of compound 1 (10.5 g, 65%); FAB MS [M+1]+ 215.3. The oxime (10 g, 46.67 mmol) was dissolved in absolute EtOH (100 mL) followed by the addition of Raney Ni (29 g, washed with absolute EtOH). The mixture was hydrogenated in a Parr shaker at 50 psi overnight. After reaction was complete, the Raney Ni was filtered off (caution; risk of fire) and the filtrate was concentrated to give compound 2 (9.2 g, 46 mmol,98% yield) as an oil which solidified under high vacuum drying. FAB MS [M+1]+ 201.3. To a solution of the bromoacetamide derivative 3 (3.0g,6.2 mmol) (prepared in Example 3) in CH2Cl2 (62 mL) at -10 C were added Huenig's base (1.2 mL, 6.82 mmol) and compound 2 (2.48 g, 12.39 mmol). The solution was gradually warmed to RT overnight. After reaction was complete, CH2Cl2 (300 mL) was added and the mixture was washed with brine (100 mL, 3x), dried over MgSO4 and filtered. The filtrate was evaporated to dryness to give a light yellow solid which was purified by flash chromatography on silica gel (200 g), eluting with 5% [NH4OH/MeOH (1:9)] /CH2Cl2 to give a 71% yield of the title compound 4 as a white solid (2.66 g, 4.4 mmol), m.p. 78-81 C; FAB MS [M+1]+35Cl 603.1; Calcd. for C31H40N4O4Cl2, C, 61.69; H, 6.68; N,9.28; Cl,11.74. Found: C, 61.33; H, 6.94; N, 9.17; Cl, 11.27. |
| 46.4% |
With hydroxylamine hydrochloride; sodium acetate; In ethanol; at 90℃; for 12h; |
To a stirred solution of tert-butyl 4-oxopiperidine-1 -carboxylate (10.00 g, 0.0502 mol) in ethanol (100 mL) hydroxylamine hydrochloride (6.98 g, 0.100 mol) and CH3COONa (8.23 g, 0.100 mol) were added, then heated to 90 C for 12 h under nitrogen atmosphere. After completion of the reaction by LCMS, the reaction mixture was concentrated and to the crude material water (100 mL) was added followed by extraction by using dichloromethane (250 mL).The organic layer was concentrated and the crude product was purified by silica gel column (Biotage) using 18- 30% of ethyl acetate in pet ether to obtain tert-butyl-4-(hydroxyimino)piperidine-1-carboxylate (5 g, 46.4 %) as a white solid. (0830) MS: 159.1 (M+H)+- t-butyl (0831) 1H-NMR (400 MHz, DMSO- 6) d = 10.45 (s, 1 H), 3.33-3.36 (m, 4H), 2.42-2.44 (m, 2H), 2.20- 2.22 (m, 2H), 1.41 (s, 9H). |
|
With hydroxylamine hydrochloride; In pyridine; at 20℃;Molecular sieve; |
To a solution of /ert-butyl 4-oxopiperidine-l-carboxylate (50 g, 251 mmol) in pyridine (500 mL) is added molecular sieves (50 g) and the mixture is stirred at room temperature for 10 minutes, followed by the addition of NH2OH HCl (30.25 g, 427 mmol). The resulting reaction is stirred at room temperature overnight, and the reaction mixture filtered through a pad of celite to remove the molecular sieves. The filtrate is diluted with water, the layers separated and the aqueous phase extracted with more ethyl acetate. The combined organic phases are washed with brine, dried over EPO <DP n="25"/>MgSO4, filtered, and concentrated in vacuo to provide the title compound. This material is used in the next step without further purification.1H NMR (300 MHz, DMSO-ddelta): 1.52 (s, 9H), 2.36 (t, J=6.0 Hz, 2H), 2.58 (t, J=6.0 Hz, 2H), 3.50-3.58 (m, 4H). |
|
With hydroxylamine hydrochloride; sodium acetate; In ethanol; at 100℃; for 7h; |
To a solution of I-48 (50 g, 0.25 mol, 1.0 eq) in ethanol (300 mL) hydroxylamine hydrochloride (35 g, 0.50 mol, 2.0 eq) was added followed by sodium acetate (41 g, 0.50 mol, 2.0 eq) and the reaction mixture was stirred at 100 C. for 7 hours. Solids were removed by filtration and the filtrate was concentrated by evaporation. Water was added to the residue; the reaction mixture was extracted with ethyl acetate and washed with aqueous sodium bicarbonate and brine. The combined organic layers were dried on anhydrous MgSO4, the solids were removed by filtration and the filtrate was concentrated to give intermediate I-49 (54 g, 100%) as white solid that was used in the following step without further purification. MS (ESI): m/z 159.1 (M+H+). |
|
With hydroxylamine hydrochloride; sodium acetate; In ethanol; water; at 15℃; for 2h; |
To a solution of tert-butyl 4-oxopiperidine-1-carboxylate (20.0 g, 100 mmol) and sodium acetate (9.88 g, 120 mmol) in a mixture of ethanol (160 mL) and water (40 mL) was added hydroxylamine hydrochloride (8.37 g, 120 mmol) and the reaction mixture was stirred at 15 C for 2 hrs. On completion, the reaction mixture was concentrated in vacuo, and 300 mL water and 300 mL dichloromethane was added. The aqueous phase was extracted with dichloromethane (3 × 100 mL). The combined layer was dried over anhydrous sodium sulfate and concentrated in vacuo to give the title compound. 1H NMR (300MHz, CDCl3) delta = 8.71 (br. s., 1H), 3.55 (q, J = 6.8 Hz, 4H), 2.63 (t, J = 6.0 Hz, 2H), 2.35 (t, J = 6.0 Hz, 2H), 1.49 (s, 9H). |
|
With hydroxylamine hydrochloride; triethylamine; In ethanol; at 0 - 20℃; for 6h;Heating / reflux; |
Example 2 Synthesis of Compound 191 EPO <DP n="18"/>A solution of 5.8 g (0.31 mol) of N-t-Boc-piperidone in 80 ml of ethanol was cooled to O0C on an ice bath. To this was added 5.2 ml (0.04 mol) of TEA then 2.6 g (0.37 mol) of solid hydroxylamine HCl. The mixture was warmed to room temperature and then heated to reflux temperature for 6 h. The solvent was removed and the solid was partitioned between saturated aqueous ammonium chloride and ethyl acetate (100 ml each). The aqueous layer was extracted with a second portion of ethyl acetate (100 ml) and the combined extract was dried over sodium sulfate. The solvent was removed at reduced pressure to afford 6.0 g of a white solid. The solid was dissolved in 60 ml of methanol and then hydrogenated over 5% Rhodium on Alumina (0.6 g) at 5O0C. After the uptake of hydrogen ceased, the catalyst was filtered and the solvent removed to afford 5.5 g (98%) of reactant 5. The NMR spectrum was consistent with the proposed structure. |






 Chemistry
Chemistry
 Pharmaceutical Intermediates
Pharmaceutical Intermediates
 Inhibitors/Agonists
Inhibitors/Agonists
 Material Science
Material Science






 HazMat Fee +
HazMat Fee +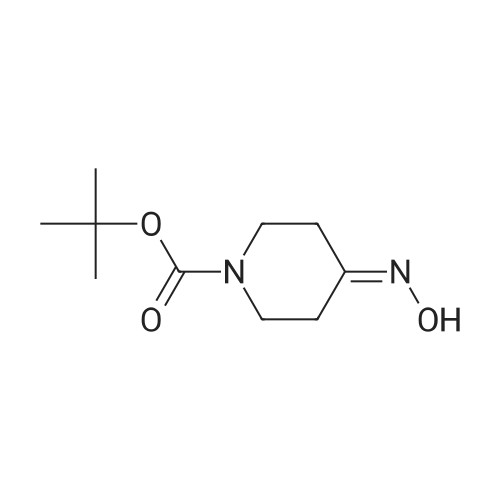

 For Research Only
For Research Only
 120K+ Compounds
120K+ Compounds
 Competitive Price
Competitive Price
 1-2 Day Shipping
1-2 Day Shipping