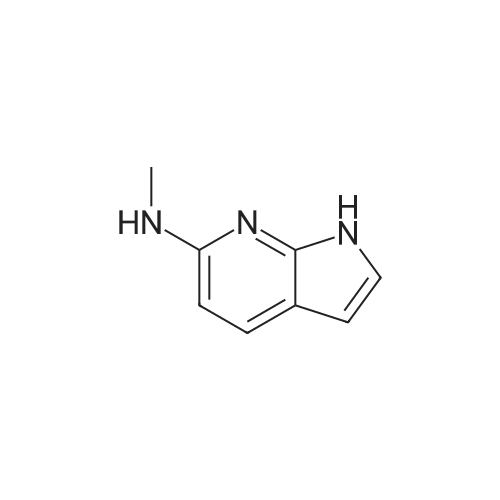
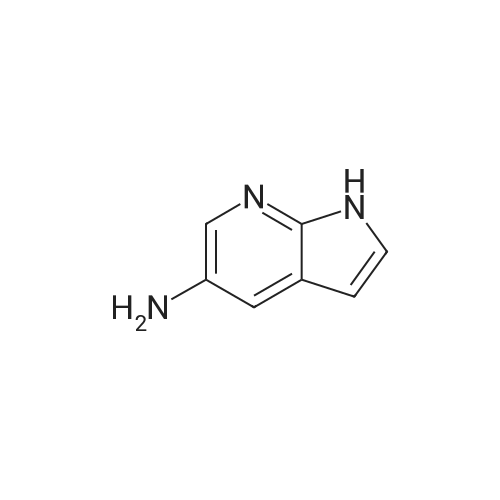
| 30% |
With sodium t-butanolate; In 1,4-dioxane; at 110℃; for 2h;Inert atmosphere; |
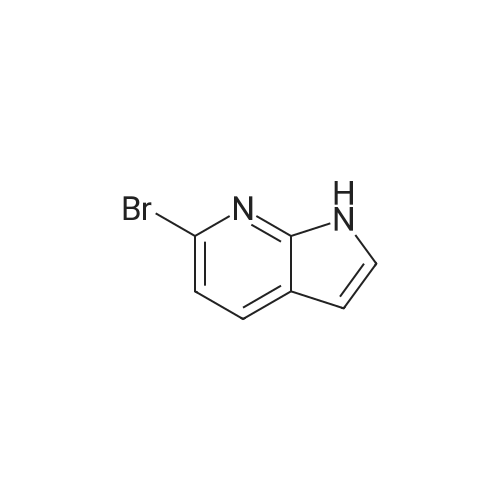
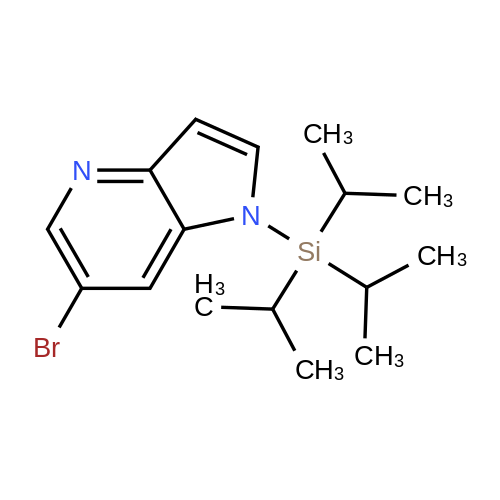
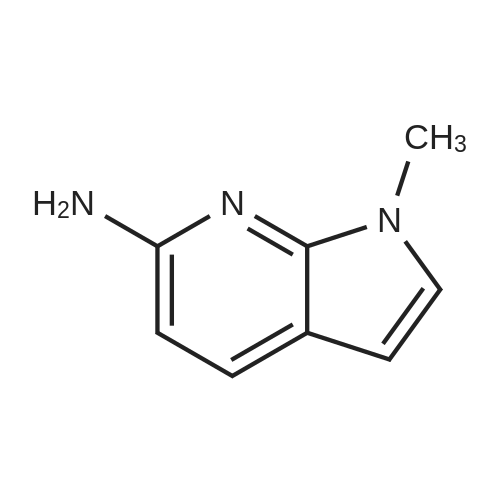
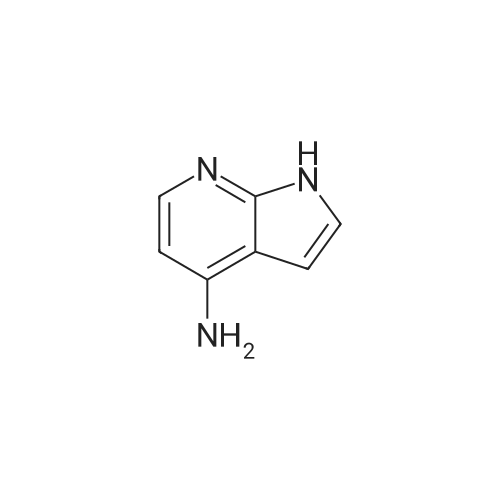
Preparative Example 60; Step AStep AAn oven-dried Schlenk flask was evacuated and back filled with argon gas. The procedure was repeated for 3-4 times. At room temperature dioxane (3 ml_) was added by a syringe and degassed by bubbling argon through the mixture. Then 2-dicyclohexylphosphino-2',4',6'- triisopropylbiphenyl (XPhos, 0.054 g, 0.1 12 mmol) and palladium(ll) acetate (0.009 g, 0.037mmol) were added together. The mixture was heated at 110 C for 1 minute. The reaction mixture became a clear red color solution. Then the title compound from Preparative Example 3 (0.050 g, 0.375 mmol), commercially available 6-bromo-1-(triisopropylsilyl)-1 /-/-pyrrolo[3,2- £>]pyridine (0.132 g, 0.375 mmol) and sodium tert butoxide (0.12 g, 1.25 mmol) were added together under an argon atmosphere. The reaction mixture was heated at 110 C for 2 h. The reaction mixture was diluted with ethyl acetate (150 mL). The organic phase was washed with water, brine, and was dried over Na2S04. The solvent was removed and the residue was purified by chromatography on silica using ethyl acetate to afford the title compound (0.045 g, 30 %). H-NMR (400 MHz, CDCI3): delta = 1.13 (d, 18H), 1.61-1.68 (m, 3H), 6.35 (dd, 1H), 6.60 (d, 1H), 6.80 (d, 1H), 6.94 (dd, 1H), 7.10 (brs, 1H), 7.39 (d, 1H), 7.73 (d, 1H), 8.20 (s, 1H), 8.54 (s, 1H), 9.27 (s, 2H) |
| 30% |
With sodium t-butanolate; XPhos;palladium diacetate; In 1,4-dioxane; at 110℃; for 2h;Inert atmosphere; |
An oven-dried Schlenk flask was evacuated and back filled with argon gas. The procedure was repeated for 3-4 times. At room temperature dioxane (3 mL) was added by a syringe and degassed by bubbling argon through the mixture. Then 2-dicyclohexylphosphino-2',4',6'-triisopropylbiphenyl (XPhos, 0.054 g, 0.112 mmol) and palladium(II) acetate (0.009 g, 0.037 mmol) were added together. The mixture was heated at 110 C. for 1 minute. The reaction mixture became a clear red color solution. Then the title compound from Preparative Example 3 (0.050 g, 0.375 mmol), commercially available 6-bromo-1-(triisopropylsilyl)-1H-pyrrolo[3,2-b]pyridine (0.132 g, 0.375 mmol) and sodium tert butoxide (0.12 g, 1.25 mmol) were added together under an argon atmosphere. The reaction mixture was heated at 110 C. for 2 h. The reaction mixture was diluted with ethyl acetate (150 mL). The organic phase was washed with water, brine, and was dried over Na2SO4. The solvent was removed and the residue was purified by chromatography on silica using ethyl acetate to afford the title compound (0.045 g, 30%).1H-NMR (400 MHz, CDCl3): delta=1.13 (d, 18H), 1.61-1.68 (m, 3H), 6.35 (dd, 1H), 6.60 (d, 1H), 6.80 (d, 1H), 6.94 (dd, 1H), 7.10 (brs, 1H), 7.39 (d, 1H), 7.73 (d, 1H), 8.20 (s, 1H), 8.54 (s, 1H), 9.27 (s, 2H) |
| 30% |
With palladium diacetate; sodium t-butanolate; XPhos; In 1,4-dioxane; at 110℃; for 2h;Inert atmosphere; |
An oven-dried Schlenk flask was evacuated and back filled with argon gas. The procedure was repeated for 3-4 times. At room temperature dioxane (3 mL) was added by a syringe and degassed by bubbling argon through the mixture. Then 2-dicyclohexylphosphino-2?,4?,6?-triisopropylbiphenyl (XPhos, 54 mg, 0.112 mmol) and palladium(II) acetate (9 mg, 0.037mmol) were added together. The mixture was heated at 110 oC for 1 minute. The reaction mixture became a clear red color solution. Then compound 5 (50 mg, 0.375 mmol), commercially available 6-bromo-1-(triisopropylsilyl)-1H-pyrrolo[3,2-b]pyridine 31 (132 mg, 0.375 mmol) and sodium tert butoxide (120 mg, 1.25 mmol) were added together under an argon atmosphere. The reaction mixture was heated at 110 oC for 2 h. The reaction mixture was diluted with ethyl acetate (150 mL). The organic phase was washed with water, brine, and was dried over Na2SO4. The solvent was removed and the residue was purified by chromatography on silica using ethyl acetate to afford the compound 38a (45 mg, 30 %). 1H-NMR (400 MHz, CDCl3): delta = 9.27 (s, 2H), 8.54 (s, 1H), 8.20 (s, 1H), 7.73 (d, 1H), 7.39 (d, 1H), 7.10 (br-s, 1H), 6.94 (dd, 1H), 6.80 (d, 1H), 6.60 (d, 1H), 6.35 (dd, 1H), 1.68-1.61 (m, 3H), 1.13 (d, 18H). |






 Chemistry
Chemistry
 Pharmaceutical Intermediates
Pharmaceutical Intermediates
 Inhibitors/Agonists
Inhibitors/Agonists
 Material Science
Material Science







 For Research Only
For Research Only
 120K+ Compounds
120K+ Compounds
 Competitive Price
Competitive Price
 1-2 Day Shipping
1-2 Day Shipping