| 95% |
With montmorillonite K10 Clay; In methanol; dichloromethane; at 38 - 42℃; |
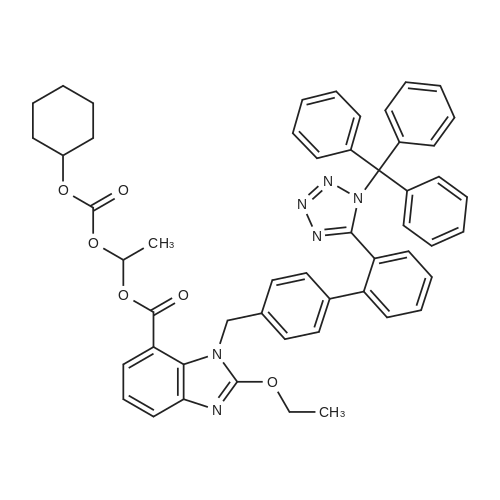
(1) 409.2 mL of dichloromethane was sequentially added to a 1 L reaction flask.Trityl candesartan cilexetil (42.6 g, 0.050 mol),136.4 mL of anhydrous methanol and 13.6 g of montmorillonite,The temperature is raised to 38 ~ 42 C reflux reaction for 4 ~ 24h,The HPLC content <3.0% of <strong>[170791-09-0]trityl candesartan cilexetil</strong> was detected by HPLC until the end of the reaction.The reaction solution is filtered,The filter cake was washed with 68.2 mL of dichloromethane.The filtrates were combined and cooled to 20-25 C.Add 409.2mL of water,The pH was adjusted to 4.5 to 5.5 with 1% dilute hydrochloric acid and stirred for 10 min.After standing for 30 min, the separated organic layer was washed twice with 409.2 mL.(2) The organic layer was evaporated to dryness under reduced pressure.Add methyl tert-butyl ether (MTBE, 341.0 mL) preheated to 35-40 C,Add a little seed crystal, cool to 15 ~ 25 C for 10 h, filter.The obtained wet product was beaten with methyl tert-butyl ether (272.8 mL) at 15 to 25 C for 3 h.Filter and filter cake was washed with methyl tert-butyl ether (68.2 mL).The wet product is vacuum dried at 50-55 C.Candesartan cilexetil 46.4 g, yield 95%.The HPLC purity was 99.4%.(3) Recovery of methyl triphenyl methyl ether:The methyl t-butyl ether solution which was filtered out and washed in the previous step was evaporated to dryness under reduced pressure.Then 200 mL of methanol was added. Cooling to -10 C to precipitate crystals,Filter, wash with methanol and dry.Add 80 mL of heptane to 16 g of dry material.Then, thionyl chloride (6.4 ml) was added dropwise at room temperature.After stirring for 10 min, it was boiled for 20 hours.Half of the solvent was distilled off and the residue was stirred at room temperature for 6 hr then at 0 C overnight. The trityl chloride is filtered off and washed with heptane and used as it is.80% recovery of trityl derivatives from undeprotected starting materials |
| 94.6% |
|
EXPERIMENT 6 (CANDESARTAN CILEXETIL)50 g of <strong>[170791-09-0]trityl candesartan cilexetil</strong> are dissolved in a mixture of 145 ml of dichloromethane and 125 ml of methanol. The solution is cooled to approximately 50C and a solution of 7.6 ml of methanesulfonic acid in 25 ml of methanol is added within 15 to 20 min. The mixture is stirred at approximately 3C for 60 min. The reaction mixture is then added to a mixture of 100 ml of dichloromethane, 190 ml of water and 88 ml of satrated NaHCO3 solution The pH ofthe mjxtture is adjusted to a pH of 6.4 to 6.5 with approximately 15 ml of saturated NaHCO3 solution and the mixture is stirred for approximately 15 min. Layers are separated and the aqueous layer is extracted with 100 ml of dichloromethane. The combined dichloromethane layers are separated and extracted with IOO ml of water. The solution is concentrated in vacuo to approximately I08 g. 100 ml of acetone are added and the mixture is again concentrated in vacuo to about 100 g. 15 ml of ethanol are added to the residue. Seeds of candesartan cilexetil are added and the suspension is stirred for approximately 3 hours at ambient temperature. 7. 5 ml of ethanol are added, the suspension is stirred for 1 hou.r and is then stored at 40C overnight. The suspension is warmed to room temperature and 350 ml of heptane are slowly added within 40 EPO <DP n="13"/>min. The suspension is stirred for 1 hour at ambient temperature and then for additional 3 hours in an ice bath. The product is then isolated by filtration, washed with 125 ml of heptane and dried in vacuum over night at ambient temperature.. Yiled 31 ,56 g (94,6%). |
| 94% |
With DIAION WK60L; In methanol; dichloromethane; for 12h;Reflux;Product distribution / selectivity; |
One hundred ml of methanol and 100 ml of methylene chloride (MC) were added to 10 g of l-(cyclohexyloxycarbonyloxy) ethyl 2-ethoxy-l-((2'-(l-trityl-lH- tetrazol-5-yl) biphenyl-4-yl) methyl)-lH-benzo[d]imidazole-7-carboxylate(Pharmacostech), then to the reaction mixture was added 10 g of weak acidic resin (DIAION WK60L, Mitsubishi Chemical Co.), followed by refluxing for 12 hours. The solid components were filtered out from the reaction mixture and washed with 100 ml of methanol. The filter-in solution was cooled to 0C and the solid formed was removed by filtration, followed by concentration under vacuum. The obtained solid substance was dissolved into a small quantity of acetone. Then n-hexane was added to the acetone solution to obtain 6.73 g (yield rate: 94%) of the standard compound represent by the following Formula 7: 1H NMR (300 MHz, DMSOd6), delta 7.80 (d, IH, >6.02 Hz), 7.61-7.74 (m, 2H), 7.46-7.57 (m, 3H), 7.24 (m, IH), 6.92-7.05 (m, 4H), 6.83 (q, IH, Jt=5.31, 5.49 Hz), 5.49 (s, 2H), 4.56-4.66 (m, 3H),1 .82 (bs, 2H), 1.62 (bs, 2H), 1.05-1.46 (m, 13H). |
| 91% |
|
EXAMPLE 3 Using 10.0 g of (+-)-1-(cyclohexyloxycarbonyloxy)ethyl 2-ethoxy-1-[2'-(N-triphenylmethyl-1H-tetrazol-5-yl)biphenyl-4-yl]methylbenzimidazole-7-carboxylate, the reaction and after-treatment of Example 1 was repeated except that ethanol was used in lieu of methanol. The procedure provided 6.83 g of (+-)-1-(cyclohexyloxycarbonyloxy)ethyl 2-ethoxy-1-[2'-(1H-tetrazol-5-yl)biphenyl-4-yl]methylbenzimidazole-7-carboxylate (yield 91%). In physicochemical properties, this compound was in agreement with the compound obtained in Example 1. |
| 90.5% |
|
Example 16: Deprotection without Acid; A suspension of <strong>[170791-09-0]cilexetil trityl candesartan</strong> (50.0 g, 58.62 mmol), water (2.64 g, 2.5 eq), and methanol (500 ml, 10 eq. by volume) was refluxed for about 16. 5 h to obtain a clear solution. The solvents were removed by evaporation at 30 mbar and 40C to obtain a solid residue (51. 7 g). The residue was dissolved at 60 C in a mixture of toluene/methanol (95: 5 w/w, 125 g), cooled to 20-23 C and stirred for about 15 h. A precipitate appeared and was collected by filtration, washed with a cold (4 C) mixture of toluene/methanol (95: 5 w/w, 25 g), and dried for 2 h at 50C and 30 mbar to give a crude solid candesartan cilexetil (32.41 g, 90.5 %), with 99.32% purity by HPLC. |
| 86.8% |
|
Example 17: Deprotection without Acid; A solution of Trityl Candesartan Cilexetil (100.0 g, 0. 117mol), Water (5.3 g), Toluene (600 mL) and Methanol (600 mL) was refluxed for about 10 h (HPLC in process control) and the solvents were evaporated at 60 C/30 mbar to obtain an oily residue. A part from the residue (6.84 g) was dissolved at 50 C in a mixture of Toluene/Methanol 95 : 5 (w/w), (11.2 g). A solution was stirred for about 6 h at 2-8'C, the solids were filtered off, washed with a cold mixture of Toluene/Methanol 95: 5 (w/w), (3.4 g) and dried at 60 C/30 mbar to the constant weight to give white solid (3.47 g, 86.8 %), 99.15 % pure by HPLC. |
| 84% |
|
Step 2: Preparation of candesartan cilexetil A mixture of N-<strong>[170791-09-0]trityl candesartan cilexetil</strong> (120 gm) in dichloromethane (720 mL) and methanol (360 mL) was taken in a round botton flask at ambient temperature followed by addition of formic acid (120 gm) over 5 minutes. The reaction mixture was heated to 30 C to 35 C and stirred at the same temperature for 7 hours. The reaction mixture was cooled to 15 C, and ethyl acetate (480 mL) and de-ionized water (480 mL) were charged to the reaction mixture; this was followed by pH adjustment with 10% aqueous sodium bicarbonate (1750 mL) at 15 C to 20 C. Ethyl acetate (120 mL) and de-ionized water (120 mL) were charged to the reaction and stirred for 20 minutes at the same temperature. The aqueous layer was extracted with ethyl acetate (2 X 240 mL) and ethyl acetate (240 mL) was added to the combined organic layer which was further washed with 20% aqueous sodium chloride. The organic layer was concentrated completely under vacuum at 25 C to 30 C and methyltertiarybutyl ether (120 mL) was added to remove the traces of ethyl acetate at the same temperature. Methyltertiarybutyl ether (480 mL) was added to the reaction mixture, stirred for 4 hours to 5 hours at ambient temperature, and cooled to 8 C to 12 C. The cooled reaction mixture was further stirred for 8 hours 12 hours, solid were filtered. The reaction mixture was washed with pre-cooled methyltertiarybutyl ether(120 mL) at 8 C to 12 C and dried under vacuum for 1 hour and further dried for 16 hours at 35 C to 40 C.Yield: 84%Chromatographic purity: 99.24%Desethyl candesartan cilexetil- 0.05 %Step 3: Purification of candesartan cilexetilCrude candesartan cilexetil (50 gm) was suspended in acetone (350 mL) at ambient temperature and stirred for 30 minutes at the same temperature. The resulting solution was filtered to remove insoluble particles and the obtained filtrate was warmed to 35 C to 40 C and de-ionized water (150 mL) was added at the same temperature. The resulting mass was slowly cooled to 2 C to 6 C in 1.5 hours and further stirred for 5 hours at the same temperature. The solid obtained was filtered, washed with chilled mixture of acetone:de-ionized water (2 X (35+35 mL)) and dried under vacuum at 35 C to 40 C to give pure candesartan cilexetil.Yield: 85% Chromatographic purity: 99.77%Desethyl candesartan cilexetil -NDThe candesartan cilexetil (30 gm) obtained above was heated in methanol (270 mL) in a round bottom flask at 35 C to 40 C and then cooled to 13 C to 17 C in 1 hour. The reaction mixture was stirred further for 7 hours and the solid was filtered and further washed with chilled methanol (12 mL). This was finally dried under vacuum at 35 C to 40 C for 16 hours.Yield : 88%Chromatographic purity: 99.90%Desethyl candesartan cilexetil -NDEthyl candesartan-0.076% |
| 83 - 94% |
With methanol; water; In toluene; for 4 - 4.5h;Heating / reflux;Product distribution / selectivity; |
Example 1 A solution of <strong>[170791-09-0]trityl candesartan cilexetil</strong> (70 g, 82 mmol), toluene (210 ml), methanol (420 ml), and water (3.5 ml) was refluxed for about 4.5 hours. The clear solution was cooled, filtered, and the filtrate was returned to the reactor. Water (350 ml, 5 ml/g <strong>[170791-09-0]trityl candesartan cilexetil</strong>) was added, and the solution was stirred for a few minutes, giving two liquid phases after the mixing was stopped. The bottom phase (toluene, 225.5 g) was collected to a vessel, while the upper phase was left in the reactor. Toluene (70 ml, 1 ml/g <strong>[170791-09-0]trityl candesartan cilexetil</strong>) was added to the upper phase, and the solution was stirred for a few minutes, giving two liquid phases after the mixing was stoppedThe reactor was emptied from the methanol-water phase (now in the bottom), the toluene phase (80 g) was added to the first toluene phase, and the combined phase was returned to the reactor. The reactor was cooled to 0C, stirred for 16 hours, and filtered. The solids were washed with toluene (1 ml/g <strong>[170791-09-0]trityl candesartan cilexetil</strong>) to give 43.5 g on dry basis. Yield: 86% by weight; Example 2 EPO <DP n="7"/>A solution of trityl candosartan cilexetil (TCS, 70 g, 82 mmol), toluene (210 mL), methanol (420 mL) and water (3.5 mL) was refluxed for about 4 h. The clear solution was cooled and filtered. The filtrate was returned to the reactor and 2 volumes of water were added (140 mL). The solution was stirred for a few minutes, giving two liquid phases after the mixing was stopped. The bottom phase (toluene, 220.5 g) was collected to a vessel, while the upper phase was left in the reactor. 1 volume of toluene (70 mL) and 1 volume of water (70 mL) were added to the upper phase. The solution was stirred for a few minutes, giving two liquid phases after the mixing was stopped.The reactor was emptied from the methanol-water phase (now in the bottom) and the toluene phases (52 g) were added to the first toluene phase, and the combined phase was returned to the reactor. The reactor was cooled to 00C, stirred for 17 hours, and filtered. The solids were washed with 1 volume of toluene to give 47.5 g on dry basis. Yield: 94 % by weight; Example 3 A solution of <strong>[170791-09-0]trityl candesartan cilexetil</strong> (TCS, 50 g, 59 mmol), toluene (150 mL), methanol (300 mL) and water (2.5 mL) was refluxed for about 4.5 h. The clear solution was cooled and filtered. The filtrate was returned to the reactor and 2 volumes of water were added (10OmL). The solution was stirred for a few minutes, giving two liquid phases after the mixing was stopped. The bottom phase (toluene, 147 g) was collected to a vessel while the upper phase was left in the reactor. 3 volumes of toluene (150 mL) were added to the upper phase. The solution was stirred for a few minutes, giving two liquid phases after the mixing was stopped.The reactor was emptied from the methanol-water phase (now in the bottom) and the toluene phases (140 g) were added to the first toluene phase, and the combined phase was returned to the reactor. The reactor was cooled to 0C, stirred for 24 hours, and filtered. The solids were washed with 1 volume of toluene to give 27.9 g on dry basis. Yield: 83 % by weight. |
| 80% |
|
Example 15. Obtaining candesartan cilexetil Methane sulphonic acid (62 mg) is added to a solution of cilexetil 2-ethoxy-l- [2 '- (trityl) -2H-tetrazole-5- yl) [ 1, 1 ' -biphenyl] -4-yl] methyl] - lH-benzimidazole-7- carboxylate (0.42 g) , which can be obtained from candesartan according to the method described in examples EPO <DP n="23"/>7 and 8 of European patent EP 459.136 Bl, in methylene chloride, keeping the temperature between -5 and 0C. Once the acid has been added, the ice bath is removed and the mixture is stirred for 2 hours at room temperature. When 5 the reaction is completed, cold water and methylene chloride are added and the pH of the resulting mixture is adjusted to approximately 6.3 with a solution of sodium bicarbonate in water 7% (p/v) . The aqueous phase is separated and extracted with methylene chloride. The10 organic phases are combined, washed with water and concentrated at reduced pressure. Acetone is added to the concentration residue and it is concentrated again at reduced pressure. The so obtained solid is recrystallised with ethanol/hexane, filtered and dried under vacuum at15 40C. (80% yield) . |
| 78.6% |
With formic acid; In methanol; toluene; for 10h;Heating / reflux;Product distribution / selectivity; |
Example 7 Method of Deprotection using Formic Acid; A solution of <strong>[170791-09-0]trityl candesartan cilexetil</strong> (30 g, 0.035 mol) in toluene (180 ml), methanol (180 ml), and formic acid (1.6 g, 0.035 mol) was refluxed for about 10 h. The reaction was monitored using HPLC. Thereafter, the solution volume was reduced by evaporation under reduced pressure (30 mbar) at a temperature of about 55 C to 60C to obtain viscous oil (36.5 g). The oil was dissolved in a mixture of toluene: methanol (65.7g : 7.3 g), stirred at about 0C to 5C until crystallization started, and kept at 2C to 8C for about 20 hours. The solids were collected by filtration, washed on the filter with a mixture toluene/methanol (90: 10 w/w, 15 g), and dried under reduced pressure (10-50 mm Hg) at a temperature of about 50C to 55 C to yield candesartan cilexetil (16.88 g, 78.6 %) as a white powder. |
| 78.0% |
With boric acid; In ethanol; at 25 - 30℃; for 8h;Heating / reflux;Product distribution / selectivity; |
Step-3: Preparation of Cilexetil candesartan; Boric acid (9.0 g) is added to a suspension of Cilexetil trityl candesartan (wet wt, 180 g) in ethanol (1000 ml) at temperature of 25 - 300C, temperature of reaction mass is raised and maintained at reflux temperature for 8 hrs. Reaction mass is concentrated to one third of its original volume by distillation of solvent and cooled the solution to 25 - 3O0C. n-Hexane (500 ml) is added to the reaction mass, stirred for 8hrs at 25 - 3O0C and filtered the product. Wet cake is washed with n-hexane (100 ml) and dried the material at a temperature of 45- 5O0C till constant weight. Dry weight of Cilexetil candesartan is 70 g (Yield: 78.0%); Example 2: Preparation of Candesartan cilexetil (without isolation of <strong>[170791-09-0]cilexetil trityl candesartan</strong>); Carbohexyl 1-chloroethyl carbonate (36 g) is added to a suspension of trityl candesartan (100 g), potassium carbonate (24 g) and potassium iodide (12 g) in DMSO (500 ml) at <n="8"/>temperature of 60 - 65C over 30 min. Reaction mass is maintained at 60-650C for 2 hrs, added toluene (300 ml) and water (300 ml). Reaction mass is mixed for 15 min., allowed to settle, the layers are separated at 60 - 650C and aqueous layer is extracted with toluene (200 ml). Water (200 ml) washings are given to the combined organic layer and toluene extractions twice at temperature of 60 - 650C. Toluene is distilled off from water washed organic layer at temperature below 6O0C under vacuum, ethanol (100 ml) is added, mixed for about 30 min and distilled off solvents under vacuum at temperature below 6O0C under vacuum. Residue is cooled to 30 - 350C, ethanol (1000ml) and boric acid (9.0 g) is added at temperature of 25 - 300C, temperature of reaction mass is raised and maintained at reflux temperature for 8 hrs. Reaction mass is concentrated to one third of its original volume by distillation of solvent and cooled the solution to 25 - 3O0C.n-Hexane (500 ml) is added to the reaction mass, mixed for 8hrs at 25 - 3O0C and filtered the product. Wet cake is washed with n-hexane (100 ml) and dried the material at temperature of 45-500C till becomes constant weight. Dry weight of Cilexetil candesartan is 65 g (Yield: 72.5%) |
| 78% |
With tin(ll) chloride; In methanol; dichloromethane; for 4h;Heating / reflux;Product distribution / selectivity; |
Step 5: Detritylation; Reaction vessel is charged with tritylcandesartan cilexetil (21.0 g, 24.6 mmoi), methanol (63 mi), dichioromethane (250 ml), anhydrous tin (IS) chloride (3.35 g, 17.6 mmol) and heated at reflux temperature for 4h. Mixture is cooled to 3O0C, water (250 ml) and 1.57 g oxalic acid are added and the precipitate is separated. After that phases in filtrate are separated and organic phase is washed with water (2x250 ml). Organic phase is dried with magnesium sulphate (18 g) and then filtered and evaporated to oily residue (22 g), which is then dissolved in isopropyl acetate (60 ml) and stirred at room temperature for 3 h. Precipitate formed is filtered off and dried in vacuum drier at 45C for 2 h. Partly dried product candesartan cilexetil (1 1.7 g, 78%) is suspended for 1 h in terf-butyl methyl ether (107 ml), then filtered off and dried over night at room temperature. |
| 74% |
In methanol; toluene; at 60 - 70℃; for 19h;Product distribution / selectivity; |
Example 10: Deprotection without Acid; Cilexetil trityl candesartan (5.0 g, 5.86 mmol) was dissolved at 60C in toluene (30 ml). Methanol (30 ml) was added and the solution was heated in an oil bath to 70C for about 19 h. The volume of the solution was reduced at 50C to 60C under reduced pressure to a weight of about 16 g and then cooled to-10C for about 48 h. The precipitated solids were collected by filtration, washed with cold methanol (MeOH at about 0C to 5C ; 2 ml x 2), dried on the filter for about 1 h to give crude candesartan cilexetil (3.1 g, 88.5%). The crude candesartan cilexetil was dissolved at reflux in methanol (23 ml), the solution was filtered under reduced pressure, and cooled under stirring in an ice bath for about 3 h. White solids were collected by filtration, washed with methanol (2.5 ml x 3), and dried in the open air overnight to give candesartan cilexetil as a white solid (2.3 g, 74 %) with 99. 28% purity by HPLC. |
| 72% |
|
A mixture of cilexetil trityl Candesartan in MDC was cooled at -10 to -5 C. A mixture of, methanol and hydrochloric acid was added to the reaction mixture at -10 to -5 C. and maintained for 3 hours. Sodium bicarbonate solution was added to the reaction mixture and organic layer was separated. Aqueous layer was extracted with MDC. Both organic layers were combined and washed brine solution. MDC was distilled out under vacuum to give residue. A mixture of rectified spirit and cyclohexane was added to the residue and stirred for 3 hours. The mixture was filtered and washed with mixture of rectified spirit and cyclohexane. The solid was dried to obtain Candesartan cilexetil.Purification of Crude Candesartan CilexetilA mixture of crude candesartan cilexetil, acetone and water was stirred at 55-60 C. The hot solution was filtered and filtrate was cooled at ambient temperature for 3 hours. The mixture was filtered and washed with mixture of acetone and water. The solid was dried to obtain pure Candesartan cilexetil.Yield: 68-72% |
| 68 - 72% |
|
Examples-5; Preparation of Candesartan cilexetil A mixture of cilexetil trityl Candesartan in MDC was cooled at -10 to -50C. A mixture of methanol and hydrochloric acid was added to the reaction mixture at -10 to -50C and maintained for 3 hours. Sodium bicarbonate solution was added to the reaction mixture and organic layer was separated. Aqueous layer was extracted with MDC. Both organic layers were combined and washed brine solution. MDC was distilled out under vacuum to give residue. A mixture of rectified spirit and cyclohexane was added to the residue and stirred for 3 hours. The mixture was filtered and washed with mixture of rectified spirit and cyclohexane. The solid was dried to obtain Candesartan cilexetil.Purification of crude candesartan cilexetil A mixture of crude candesartan cilexetil, acetone and water was stirred at 55-600C. The hot solution was filtered and filtrate was cooled at ambient temperature for 3 hours. The mixture was filtered and washed with mixture of acetone and water. The solid was dried to obtain pure Candesartan cilexetil.Yield: 68-72 % |
| 66.4% |
With formic acid; In methanol; dichloromethane; at 25 - 27℃; for 5h;Product distribution / selectivity; |
Example 5: Method of Deprotection using Formic Acid; A solution of <strong>[170791-09-0]cilexetil trityl candesartan</strong> (2.0 g, 2.35 mmol), formic acid (2.16 g, 46.9 mmol), dichloromethane (8 ml) and methanol (4 ml) was stirred at 25 C to 27C for about 5 h (TLC monitoring). The reaction mixture was neutralized with a saturated solution of sodium bicarbonate. The dichloromethane was removed under reduced pressure, the residue was diluted with water (10 ml) and extracted with ethyl acetate (20 ml x 2). The combined organic layer was washed with brine (10 ml x 2), dried over sodium sulfate and evaporated to give an oil (2.05 g) which was crystallized from tert- butyl methyl ether (TBME) (2.7 g) to give Candesartan cilexetil (0.95 g, 66. 4 %). |
| 60% |
With formic acid; In methanol; dichloromethane; at 30 - 35℃; for 5h; |
To a mixtureof 1- (cyclohexyloxycarbonyloxy) ethyl 2-ethoxy-1- [ [2'- (N-triphenyl methyltetrazol-5-yl) [1, 1'-biphenyl] -4-yl] methyl]-lH-benzimidazole-7-carboxylic acid of Formula II (B) (50.0 gm) in methylene chloride (50.0 ml) and methanol (75.0 ml) was added formic acid (50.0 ml, 99%) slowly under stirring while maintaining the temperature below35 C. The resultant mass was stirred at30-35 C for further 5 hours. After completion of reaction, the reaction mass was cooled to about20 C and charged into a pre-cooled mixture of ethyl acetate (500.0 ml) and water (250.0 ml). The resultant reaction mixture was stirred for 30 minutes and layers were separated. The aqueous layers were combined and extracted with fresh ethyl acetate (125.0 ml). The combined organic layers were washed with sodium bicarbonate solution (10%,500. 0 ml) and finally with water (50.0 ml). The organic layer was dried over anhydrous sodium sulphate and to it was added activated carbon (2.0 gm, 4% w/w). After stirring for 30 minutes at20-25 C, it was filtered through a celite bed and the bed was washed with ethyl acetate (50.0 ml). The filtrate was concentrated to dryness under vacuum and at a temperature not exceeding 35-40 C. To the residue was added cyclohexane (500.0 ml) and the resultant mixture was stirred at25-30 C for 12 hours. The separated product was filtered and after washing with cyclohexane (50.0 ml), it was dried in a vacuum oven at a temperature of about35-40 C to afford crude candesartan cilexetil, which after first purification from acetone-water followed crystallization by from methanol gave pure title compound. Yield: 28. 0 gm (60%) HPLC Purity: 99.5% |
|
With methanesulfonic acid; In methanol; dichloromethane; at 25 - 27℃; for 4h;Product distribution / selectivity; |
Example 3: Method of Deprotection using Methanesulfonic Acid; A solution of <strong>[170791-09-0]cilexetil trityl candesartan</strong> (0.50 g, 0.59 mmol), metlianesulfonic acid (0.09 g, 0. 88 mmol), dichloromethane (10 ml) and methanol (1 ml) was stirred at 25 C to 27C for about 4 h. The reaction was monitored using thin layer chromatography (TLC monitoring). The reaction mixture was neutralized with a saturated solution of sodium bicarbonate and the dichloromethane was removed under reduced pressure. The residue was diluted with water (10 ml) and extracted with ethyl acetate (20 ml x 2). The combined organic layers were washed with brine (10 ml x 2), dried over sodium sulfate, and evaporated to give Candesartan cilexetil crude. |
|
With trifluoroacetic acid; In methanol; toluene; at 20 - 25℃; for 6.83333 - 21.5h;Product distribution / selectivity; |
Example 8: Method of Deprotection using Trifluoroacetic Acid; The protecting group (trityl) was removed using strong organic acids. Trifluoroacetic acid (0.1 ml, 1.3 eq. ) was added at 20C to 25 C to a stirred suspension of <strong>[170791-09-0]cilexetil trityl candesartan</strong> (1 g) in methanol (6 ml) and toluene (6 ml). After 50 min of stirring at 20C to 25 C a solution formed. The solution was stirred at 20C to 25 C for about an additional 6 h. Thereafter, the pH of the solution was adjusted to 6.4 with a saturated aqueous solution of sodium bicarbonate, the solution was diluted with brine (20 ml), and extracted with ethyl acetate (20 ml x 2). The combined organic layers were washed with brine (10 ml), dried over sodium sulfate, filtered, and the volume reduced by evaporation to give a semi-solid mass crude candesartan cilexetil. Alternatively, trichloroacetic acid may be used with this process.; Example 9: Method of Deprotection using Trifluoroacetic Acid; Trifluoroacetic acid (0.17 g, 0.65 eq. ) was added dropwise at a temperature of about 20C to about 25C to a stirred suspension of <strong>[170791-09-0]cilexetil trityl candesartan</strong> (2 g, 2.34 mmol) in toluene (12 ml) and methanol (12 ml). A solution formed after 1.5 h of stirring and the solution was stirred for about 20 h at a temperature of about 20C to about 25C. t Thereafter, the pH of the solution was adjusted to a pH 6.5 with a saturated solution of sodium bicarbonate, diluted with water (30 ml), and extracted with ethyl acetate (20 ml x 4). The organic layers were collected and dried over sodium sulfate, filtered, and the solvent was removed by evaporation to yield semi-solid candesartan cilexetil. |
|
With water; In methanol; toluene; for 5 - 12h;Heating / reflux;Product distribution / selectivity; |
Example 12: Deprotection without Acid; A mixture of <strong>[170791-09-0]trityl candesartan cilexetil</strong> (20 g, 23.45 mmol), toluene (60 ml), methanol (60 ml), and water (1 ml) was gently refluxed for about 12 h. The reaction was monitored by HPLC. The solution volume was reduced by evaporation under reduced pressure (30 mbar) at a temperature of about 55C 60C to obtain viscous oil of candesartan cilexetil as a residue (36.5 g). |
|
With water; In methanol; for 16 - 17h;Heating / reflux;Product distribution / selectivity; |
Example 14 : Deprotection without Acid; A mixture of <strong>[170791-09-0]trityl candesartan cilexetil</strong> (20 g, 23.45 mmol), methanol (200 ml), and water (1 ml) was gently refluxed for about 16-17 h. The reaction progress was monitored by HPLC. The solution volume was reduced by evaporation under reduced pressure (30 mbar) at a temperature of 55C to 60C to obtain viscous oil of candesartan cilexetil as a residue. |
|
With formic acid; In methanol; toluene; at 50 - 55℃; for 7h;Product distribution / selectivity; |
Example 6: Method of Deprotection using Formic Acid; A solution of <strong>[170791-09-0]cilexetil trityl candesartan</strong> (1.0 g, 1.18 mmol) was dissolved in toluene (10 ml) at 50C to 55C followed by addition of formic acid (1.1 g, 23.88 mmol), and methanol (6 ml). The solution was heated to 50C to 5. 5"C for about 7 h. The reaction mixture was cooled to 20C to 25 C, pH adjusted to pH of 6.4 with 1 N NaOH, and extracted with ethyl acetate (20 ml x 3). The combined organic layers were washed with brine (10 ml x 2), dried over sodium sulfate and evaporated to give a semi-solid mass (0.79 g). |
|
With pyridinium p-toluenesulfonate; In methanol; dichloromethane; at 25 - 27℃; for 20h;Product distribution / selectivity; |
Example 4: Method of Deprotection using p-Toluene Sulphonic Acid; A solution of <strong>[170791-09-0]cilexetil trityl candesartan</strong> (0.50 g, 0.59 mmol), PPTS (pyridine para-toluene sulphonic acid, 0.22 g, 0.88 mmol), dichloromethane (10 ml) and methanol (1 ml) was stirred at 25C to 27C for about 20 h. The reaction progress was monitored using thin layer chromatography (TLC monitoring). The reaction mixture was neutralized with a saturated solution of sodium bicarbonate. The dichloromethane was removed under reduced pressure, the residue was diluted with water (10 ml) and extracted with ethyl acetate (20 ml x 2). The combined organic layers were washed with brine (10 ml x 2), dried over sodium sulfate, and evaporated to give Candesartan cilexetil crude. |
|
|
Example 11: Deprotection without Acid; Cilexetil trityl candesartan (20 g, 23 mmol) was dissolved at 60C in toluene (120 ml). Methanol (120 ml) was added and the solution was heated in an oil bath at about 75C to 80C for about 13 hours. The solution was reduced in volume by evaporation at 50C to 60C under reduced pressure to give a viscous residue (about 27 g) which was dissolved in methanol (60 ml) and the solvent removed by evaporation to dryness to give a foam (about 23 g). The foam was dissolved in methanol (about 40 g) at reflux temperature. The solution was then filtered under reduced pressure, cooled to 4C to obtain a solid and kept at this temperature for 12 to 15 hours. The precipitated solids were collected by filtration, washed with the cold methanol at about 0C to 5C (20 ml x 2) and dried at 50C under vacuum to give candesartan cilexetil (15.5 g). Trituration of candesartan cilexetil (1 g) with toluene (5 ml) at 25 C to 27C during 1 h gave candesartan cilexetil (about 0.65 g). |
|
|
A solution of 300 ml of methanol and 7,6 ml of methanesulfonic acid is cooled to approximately 3C. 50 g of <strong>[170791-09-0]trityl candesartan cilexetil</strong> is added and the mixture is stirred at approximately 3C for 30 minutes. 24.4 ml of triethylamine are added at a rate that temperature does not exceed 10C. 1000 ml of heptane are added followed by 50 ml of H2O. The mixture is stirred for 10 min in an ice bath. The layers are then separated, the lower phase is extracted with 250 ml of heptane under ice cooling. The layers are again separated and the lower phase is warmed up to approximately 20C. 1 5 ml of 2 m HCl is added drop wise followed by seeds of candesartan cilexetil. The suspension is briefly stirred and 14.3 ml of 2 m HCl are added within approximately 60 min. The suspension is then stirred for additional 30 min and the product is then isolated by filtration, washed with 75 ml of a mixture methanol and H2O ( 3:1 v/v) and dried in vacuum at ambient temperature over night. |
|
|
EXPERIMENT 1 (CANDESARTAN CILEXETIL))3 g (0,0035 mol) of trityl candersartan cilexetil was dissolved in a mixture of 9 ml of dichloromethane and 9 ml of methanol. Then 0,1 ml of water and 0,02 ml (cca one drop) of methanesulphonic acid (MSK) was added. Reaction mixture was stirred at room temperature for one hour 30 minutes, then a mixture of 6,3 ml of dichloromethane and 12,12 ml of water was added. pH was adjusted to 6,3 with a saturated solution of sodium hydrogen carbonate, organic layer was concentrated to the rest of 6,4 g. Then 6,3 ml of acetone was added and evaporated to drieness.1 ml of ethanol was added to precipitate crystals. To the resulting mixture 22 ml of n- hexane were added and stirring was continued at room temperature for one hour. The separated crystals were filtered and washed with 10 ml of a mixture of ethanol : n-hexane = 1 :9. Product was dried at 6O0C to obtain raw product (1 ,98 g) which was crystallised from 19,8 ml of i-propanole to obtain pure candesartan cilexetil in Form 1. Yield: 1 ,64 g. |
|
|
EXPERIMENT 4 (CANDESARTAN CILEXETIL)3 g (0,0035 mol) of <strong>[170791-09-0]trityl candesartan cilexetil</strong> was dissolved in a mixture of 9 ml of dichloromethane and 9 ml of methanol then 0,1 ml of water and 0,036 ml of trifluoroacetic acid was added. Reaction mixture was stirred at room temperature for one hour and 30 minutes, then a mixture of 6,3 ml of dichloromethane and 12,12 ml of water was added. pH was adjusted to ph 6,3 with a saturated solution of sodium hydrogen carbonate, organic layer was concentrated to the rest of 6,4 g. Then 6,3 ml of acetone was added and evaporated to dryness. 1 ml of ethanol was added to precipitate crystals. To the resulting mixture 22 ml of n-hexane were added and stirring was continued at room temperature for one hour. The separated crystals were filtered and washed with 10 ml of a mixture of ethanol : n-hexane = 1 :9. Product was dried at 600C to obtain raw product (2,25 g), which was crystallized from 22,5 ml of i-propanol to obtain pure candesartan cilexetil in Form 1. Yield: 1 ,87g |
|
|
Reactor cooled bellow 5 0C is charged with 85,3 kg MeOH and 0,8 L water and 3,2 kg cone sulfuric acid, or equivalent of HCI, whereto above product is added. The suspension is mixed until the completion of the reaction. Thereafter the temperature is kept bellow 10 0C and 8,5 kg Threeethylamine and 21 ,6 L water are added. Product is washed with heptane and to the methanolic phase water is added, heated to 40-45 0C and upon cooling candesartan cilexetil is crystallized. A small amount of sulfuric acid may be added during the crystallization. |
|
With methanol; water; for 3 - 4h;Heating / reflux; |
A solution of Trityl Candesartan Cilexetil (TCS, 1000 g, 1172 mmol), Toluene (3000 mL), Methanol (6000 mL) and Water (50 mL) was refluxed for about 3-4 h (HPLC control), the solvents were evaporated at 50C under reduce pressure to give a residue as a viscous oil. The residue was dissolved at 500C in a mixture of Toluene/Methanol (2960 g, 95:5, w/w). The mixture was then cooled to (-5)C to (5)C and kept at this temperature for about 12 h. The precipitated solids were filtered off, washed on the filter with cold Toluene (1000 mL) and then dried at 600C under reduced pressure to give crude candesartan cilexetil Form I (-600 g L.O.D = 17% ). |
|
With methanol; water; zinc(II) chloride; In dichloromethane; for 5h;Heating / reflux;Product distribution / selectivity; |
Example 2A mixture of 1.55 g (1.8 mmol) of <strong>[170791-09-0]trityl candesartan cilexetil</strong>, 5.4 ml of methanol, 22 ml of methylene chloride, 0.05 g (1.61 mmol) of ZnCl2 and 0.5 ml of water is stirred under reflux temperature for 5 h. The reaction mixture is analyzed (Area % HPLC: candesartan cilexetil: 76.3%, <strong>[170791-09-0]trityl candesartan cilexetil</strong>: 1.8 %, 2-oxo candesartan cilexetil: 0.7 %, ethyl ester of candesartan 0.09 %.) and cooled to room temperature. Then, to the mixture 36 ml of methylene chloride and 55 ml of water is added. The phases were separated and organic phase is washed with 2 x 55 ml of water. Organic phase is dried over Na2SO4, filtered and evaporated to the oily residue. This residue is dissolved in 1.6 ml of methylene chloride and then 16 ml of isopropyl acetate is added. The mixture is stirred at 0 0C for 24 h. The precipitated product is collected by filtration and dried. After that the product was suspended in 5 ml of tert-butyl methyl ether. The mixture is stirred for 2h. The product is collected by filtration and dried at 400C for 2h in vacuum drier (0.7 g) . Area % HPLC: Candesartan cilexetil: 99.6%, 2-oxo candesartan cilexetil: 0.00 %, ethyl ester of candesartan 0.08%. |
|
With water; In acetonitrile; for 6 - 15h;Heating / reflux;Product distribution / selectivity; |
Example 9; Candesartan cilexetil (VI); A mixture of <strong>[170791-09-0]trityl candesartan cilexetil</strong> (IV; 2 g), acetonitrile (20 ml), and water (5 ml) was refluxed for 9 h. Then, 15 ml was distilled off from the reaction mixture over 1 h, the concentration residue was cooled, and toluene (15 ml) was added. The lower layer was separated, the upper organic layer was concentrated by evaporating 12 ml of the solvent, and the mixture was cooled and allowed to stand in a refrigerator overnight. The precipitated portion was sucked off and dried in vacuo at 40 0C. 1.4 g of a white substance with an HPLC purity of 98.2 % was obtained. After crystallization from toluene a product was obtained having the meting point of 160-163 C (decomp.) and HPLC purity of 99.2 %. 1H NMR (250 MHz, CDCl3) delta: 1.13-1.50 (12H, m); 1.64 (2H, m); 1.79 (2H, m); 4.10-4.50 (3H, m); 5.62 (2H, d); 6.65-6.93 (7H, m); 7.27-7.28 (IH, m); 7.46-7.48 (IH, m); 7.56-7.59 (2H, m); 7.98- 8.02 (IH, m).; Example 10; Candesartan cilexetil (VI); A mixture of <strong>[170791-09-0]trityl candesartan cilexetil</strong> (IV; 2 g), acetonitrile (20 ml), and water (5 ml) was refluxed for 9 h. Then, 10 ml was distilled off from the reaction mixture over 1 h, the concentration residue was cooled, and ethyl acetate (10 ml) was added. 25 ml of the solvent was distilled off from the mixture, the same amount of ethyl acetate was added, and the mixture was cooled. The lower layer was separated, the upper organic layer was evaporated, the evaporation residue was dissolved hot in toluene (9 ml), and the solution was cooled and allowed to stand in a refrigerator overnight. The precipitated portion was sucked off and dried in vacuo at 40 0C. 1.5 g of a white substance with an HPLC purity of 98.7 % was obtained. After crystallization from toluene a product was obtained having the meting point of 161- 163 0C (decomp.) and HPLC purity of 99.0 %.; Example 11; Candesartan cilexetil (VI); A mixture of <strong>[170791-09-0]trityl candesartan cilexetil</strong> (IV; 2 g), acetonitrile (25 ml), and water (5 ml) was refluxed for 1O h. The reaction mixture was evaporated in vacuo, and the evaporation residue was dissolved hot in toluene (35 ml). The excluded water layer was separated, and the organic layer was concentrated to 15 ml at a normal pressure. The mixture was cooled and allowed to stand in a refrigerator overnight. The precipitated portion was sucked off and dried at 40 0C in vacuo. 1.4 g of a white substance with an HPLC purity of 98.5 % was obtained. After crystallization from cyclohexane a product was obtained having the meting point of 160- 163 C (decomp.) and HPLC purity of 99.4 %.; Example 12; Candesartan cilexetil (VI); A mixture of <strong>[170791-09-0]trityl candesartan cilexetil</strong> (IV; 2 g), acetonitrile (20 ml), and water (2 ml) was refluxed for 15 h. The reaction mixture was evaporated in vacuo, and the evaporation residue was dissolved hot in toluene (35 ml). The excluded water layer was separated, and the organic layer was concentrated to 15 ml at a normal pressure. The mixture was cooled and allowed to stand in a refrigerator overnight. The precipitated portion was sucked off and dried at 40 C in vacuo. 1.5 g of a white substance with an HPLC purity of 98.2 % was obtained. After crystallization from the mixture toluene/cyclohexane a product was obtained having the meting point of 160-163 C (decomp.) and HPLC purity of 99.4 %.; Example 14; Candesartan cilexetil (VI); A mixture of <strong>[170791-09-0]trityl candesartan cilexetil</strong> (IV; 5 g), acetonitrile (50 ml), and water (12.5 ml) was refluxed for 6 h. Then, 35 ml was distilled off from the reaction mixture over 2.5 h, the concentration residue was cooled, and toluene (35 ml) was added. The lower layer was separated, the upper organic layer was concentrated by evaporating 30 ml of the solvent, and the mixture was cooled and allowed to stand in a refrigerator overnight. The precipitated portion was sucked off and dried at 40 C in vacuo. 3.1 g of a white substance with an HPLC purity of 98.2 % was obtained. The crystals were boiled with toluene (30 ml) for 30 minutes and then stirred at room temperature overnight. A product was obtained having the meting point of 161-163 0C (decomp.) and HPLC purity of 99.5 %. |
|
With water; In acetone; for 48h;Heating / reflux;Product distribution / selectivity; |
Example 13; Candesartan cilexetil (VI); A mixture of <strong>[170791-09-0]trityl candesartan cilexetil</strong> (IV; 1 g), acetone (35 ml), and water (10 ml) was refluxed for 48 h. The reaction mixture was evaporated in vacuo, the evaporation residue was dissolved hot in toluene (20 ml), and 10 ml of an azeotropic mixture toluene/water was distilled off from the mixture. This process was repeated three times, and, after the last <n="16"/>distillation, the resulting concentrated solution was cooled and allowed to stand in a refrigerator overnight. The precipitated portion was sucked off and dried at 40 C in vacuo. 0.65 g of a white substance with an HPLC purity of 97.2 % was obtained. After crystallization from the mixture toluene/ethanol a product was obtained having the meting point of 160- 163 0C (decomp.) and HPLC purity of 99.6 %. |
|
With methanol; zinc(II) chloride; In dichloromethane; for 4h;Heating / reflux; |
17.06 g (20 mmol) of trityf candesartan cilthetaxetil, 60 ml MeOH, 240 ml methylene chloride and 2.39 g (17.5 mmol) ZnCi2 are heated under reflux for 4 hours. After the reaction is completed, the reaction mixture is cooled to approximately 300C, 200 ml of water are added, the organic and the aqueous phase are separated and the organic phase is washed with 2 x 200 ml of water and dried with Na2SO4 until the water content is below 0.3%, and the volatile components are evaporated to yield 18 g of an oily residue. The oily residue obtained is dissolved in 60 ml of isopropyi acetate and the mixture is stirred at room temperature (between 15 and 25C) for approximately 7 hours. The precipitate is filtered and dried for 2 hours at 45C. 10.7 g of a partially wet product are obtained which are further suspended in 107 ml of tert- butyl methyl ether for 1 hour at room temperature. The product is dried overnight at room temperature. |
|
With hydrogen;palladium 10% on activated carbon; In ethanol; toluene; at 20℃; under 760.051 Torr;Product distribution / selectivity; |
Example 2; Mixture of toluene (600 ml) and ethanol (300 ml) was added to 1-(Cyclo hexyloxycarbonyloxy)ethyl-2-ethoxy-1-[[2'-(N-triphenylmethyltetrazole-5-yl) biphenyl-4-yl]methyl]benzimidazole-7-carboxylate (60 gm) and hydrogenated at room temperature with hydrogen at 1 atmospheric pressure in the presence of palladium on carbon (10%, 12 gm) until the hydrogen uptake was ceased. Filtered over celite bed, washed with a mixture of toluene (100 ml) and ethanol (50 ml), filtrate was collected and concentrated below 450C. A mixture of acetone (100 ml) and n-hexane (450 ml) was added, stirred at room temperature for 2 hours, cooled to O0C and stirred for 4 hours 30 minutes, filtered, washed with a mixture of acetone (10 ml) and n-hexane (90 ml) and dried to get crude candesartan cilexetil (39 gm, HPLC purity: 92%). |
|
With methanol; iodine; at 20 - 58℃;Product distribution / selectivity; |
Trityl candesartan cilexetil (10 gm, 0.0117moles) was added to a mixture of methanol (100 mL) and iodine (0.3gm, 0.001181 moles) in a round bottom flask and the resulting solution was stirred for 15 minutes at ambient temperature to get a clear solution. The mixture was heated to 57C to 58C and stirred for 4 hours to 5 hours at the sametemperature, followed by removal of the methanol under reduced pressure, to get the residual mass. The residual mass so obtained was dissolved in dichloromethane (60mL) and washed with a 5% aqueous sodium thiosulfate (lOOmL) solution followed by washing of the organic layer with de-ionized water (40 mL). The organic layer was dried over sodium sulfate and concentrated under reduced pressure to get the residual mass. This was followed by the addition of cyclohexane (lOOmL) and subsequent stirring for 12 hours at ambienttemperature. The solid obtained was filtered, washed with cyclohexane (20mL) and air dried at 40C to get crude candesartan cilexetil.Yield: 7.45 gm |






 Chemistry
Chemistry
 Pharmaceutical Intermediates
Pharmaceutical Intermediates
 Inhibitors/Agonists
Inhibitors/Agonists
 Material Science
Material Science






 HazMat Fee +
HazMat Fee +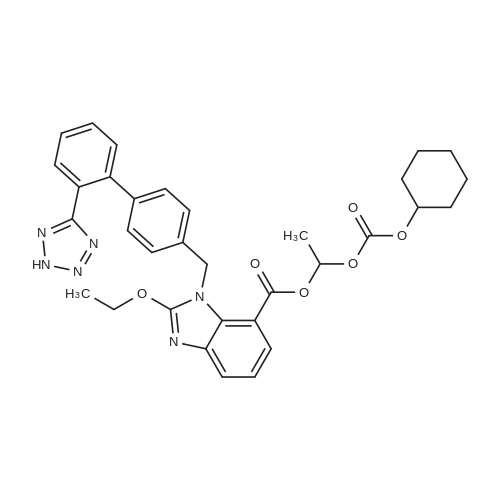

 For Research Only
For Research Only
 120K+ Compounds
120K+ Compounds
 Competitive Price
Competitive Price
 1-2 Day Shipping
1-2 Day Shipping