| 94.3% |
With sulfuric acid; In water; acetone; at 20℃; for 4.5h; |
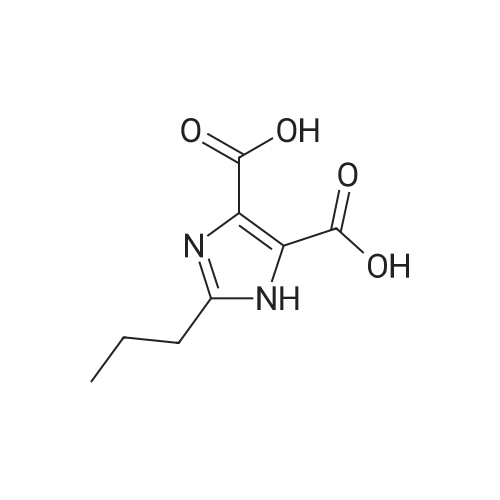
10L reaction flask was charged 500.0g (0.62mol) compound 6,3L water, 1L of acetone, was added dropwise a solution of 645.0g of 22.5% sulfuric acid at 20 . Dropping was completed, the reaction at room temperature after 4.5h TLC or HPLC in control, raw reaction was complete. Body cooling system to 0-5 , when the temperature stabilized filtrate back into the reaction vessel was charged with acetone 1L and 2L water. The system was maintained at 15 solution of 210g of potassium carbonate and 400g water to form a solution, drops of completion, stirring after 1-2h incubation filtration. The filter cake was dried to obtain olmesartan medoxomil pure 328.9g. HPLC: 99.96%, yield: 94.3% |
| 91.79% |
With acetic acid; In water; at 50℃; for 2h; |
The reaction flask was charged with 400 g of compound VII (0.50 mol) and 70% aqueous acetic acid solution of 3200 ml, Reaction was carried out at 50 C for 2 hours. After the completion of the reaction, the solvent was concentrated and concentrated, and ammonia was added to the residue.PH = 7, extracted with 1400 ml of ethyl acetate, the ethyl acetate layer was washed with water, and finally with anhydrous sodium sulfateDried; filtered, the filtrate was concentrated to dryness and the residue was recrystallized from ethanol to give the compound I pure product 256.1G, yield: 91.79%. |
| 90 - 98% |
With sulfuric acid; In water; acetone; at 20 - 40℃; for 4 - 6h;Product distribution / selectivity; |
A 250 round bottom flask was charged with MTT (10 g), acetone/water (2/2 vol.), and 3 eq of H2SO4. The combination was stirred at room temperature for about 4-6 hrs. Triphenyl carbinol (TPC) was precipitated by adding water and filtered out. NaHC03 was added to the filtrate, and the mixture was cooled to 5C and stirred for 1 hr. Crude olmesartan medoxomil was obtained as white crystals (90% yield).Example 2: Preparation of crude olmesartan medoxomil AIL reactor, equipped with mechanical stirrer and thermometer, was charged with MTT (70 g), acetone (140 ml), water (140 ml), and H2S04 (19.47 g). The reactor was heated to 40C for 2.5 hrs (at EOR, MTT is LT 1%). Water (140 ml) was added at 40C, and the reaction was stirred for 1.5 hrs or until MTT is LT 0.1%. After cooling to 15C and stirring for 1 hr, the TPC was filtered and washed with water (70 ml).NaHC03 was added in portions to the filtrate at room temperature. The reaction mixture was stirred for 1 hr, then filtrated, and the cake was washed with water (140 ml). The solid was dried at 45C in a vacuum oven overnight to obtain crude OLM-Mod (98 % yield). |
| 90% |
With water;sulfuric acid; In acetic acid; at 25 - 30℃;Product distribution / selectivity; |
A mixture of trityl olmesartan medoxomil in acetic acid and water (1:1, 400 mL) and sulfuric acid (12.2 gm) (1 mol equivalent) was stirred at 25C-30C for 45-60 minutes. Triphenylcarbinol was filtered and the filtrate was washed with acetic acid and water mixture (1:1, 50 rnL). Sodium carbonate solution (25% w/v, 100 mL) was charged to the filtrate and the product was extracted with dichloromethane (500 mL) followed by recovery of the solvent. The product was isolated, recrystallized using acetonitrile (300 mL), filtered, washed and dried under reduced pressure to obtain crude olmesartan medoxomil.Yield: 90% HPLC purity: 99.29 % OLM-acid: Not Detectable OLM- Eliminate: 0.07% Acetic acid content: Not Detectable |
| 90% |
With hydrogenchloride; water; In methanol; ethyl acetate; at 20℃; for 2h; |
To 12.0g (15 mmol) of tritylolmesartan medoxomil prepared according to examples 1 or 2 69 ml of ethyl acetate was added and after that 12 ml of methanol and 2.67 ml (32 mmol) of conc. HCl were added. The mixture was stirred at room temperature for 2h and then the mixture was cooled to below 2C. To the cooled mixture 71 ml of 1% NH3 was slowly added to adjust pH to 4.93. The phases were separated and water phase was reextracted with 20 ml of ethyl acetate. Collected organic phases were dried over Na2SO4, filtered and concentrated under reduced pressure to approximately 15g of oily residue. To this residue 15 ml of ethyl acetate were added and the mixture was cooled to 20 C and stirred at this temperature for 2h, and then cooled under 0C and stirred at this temperature for 1hour. The product was filtered off and washed with 5 ml of fresh ethyl acetate and dried. Yield: 7.55 g (90%). HPLC: 99.81 % of the product, all individual impurities under 0.10%. |
| 90.2% |
With hydrogenchloride; In water; ethyl acetate; at 15 - 20℃; for 4h; |
To the three-necked flask were added with 38 ml of ethyl acetate, 45 g of 6.4% dilute hydrochloric acid, 25 g of trityl olmesartan medoxomil was added with stirring. Stir in water bath, control the temperature 15 to 20 C, stirring about 4 hours, TLC plate detection point of raw material disappeared,That is, it is judged that the deprotection reaction is completed.Cooled to room temperature, 115 ml * 5 toluene was added to wash, the aqueous layer was added with 135 ml of acetone, potassium bicarbonate aqueous solution adjusted P=4.0, control temperature 15 ± 5 C Stir for 2 hours, Filtered and dried to give 16.0 g of olmesartan medoxomil in a yield of 90.2%. HPLC analysis: purity 98.75%, impurity a (olmesartan acid) 0.71%, individual impurities less than 0.10% |
| 89.8% |
|
Example 8; To a solution of (5-Methyl-2-oxo-1,3-dioxolen-4-yl)methyl-4-(1-hydroxy-1-methyl ethyl)-2-propyl-1-{4-[2-(trityltetrazol-5-yl)phenyl]phenyl}methylimidazole-5-carboxylate (5.0 g; 6.25 mmol) and toluene (20 ml) at 15-20 C. is added hydrochloric acid (30 ml). Reaction mass is stirred at 15-20 C. for about 15 minutes to 4 hours. The reaction is monitored by TLC. After completion of reaction layers are separated. Aqueous layer is washed with toluene (10 ml). Adjusted the pH of the aqueous layer to 5-6 using aqueous potassium carbonate solution (200 ml) and the compound is extracted using ethyl acetate (60 ml). Combined ethyl acetate layers are washed with brine. Finally crude product (3.1 g; 89.8%) is isolated by evaporation of solvent under reduced pressure.Purity: 99.5% |
| 86% |
With water; In acetone; for 14h;Heating / reflux;Product distribution / selectivity; |
Example 4; Olmesartan medoxomil (V); The starting substance (III; 10 g) was dissolved in acetone (50 ml), and, after adding water (25 g), the mixture was heated to a mild boil for 14 h. After evaporating acetone and adding ethyl acetate (50 ml), water was separated, and the organic layer was again washed with water (10 ml). The extract was concentrated and evaporated with toluene (50 ml) once more, the residue was dissolved in ethyl acetate (20 ml) and toluene (20 ml). The mixture was concentrated to 25 ml and allowed to crystallize under stirring for 30 min; after cooling to 15 C, the insoluble portion was sucked off and washed with ethyl acetate. 6.5 g of the product was obtained, which, after recrystallization from ethanol, gave 6 g (86 %) of the product with an HPLC purity of 98.7 %.By further recrystallization from ethyl acetate and cyclohexane, 5.1 g of a sample with an HPLC purity of 99.6 % was obtained. 1H NMR (250 MHz, CDCl3) delta: 0.82 (3H, t, J = 7.5 Hz); 1.50 (6H, s); 1.54-1.63 (2H, m); 2.07 (3H, s); 2.48 (2H, t, J = 7.5 Hz); 4.86 (2H, s); 5.32 (2H, s); 6.70 (2H, d, J = 8 Hz); 6.99 (2H, d, J = 8 Hz); 7.3-7.5 (3H, m); 7.72 (IH, dd, J = 1.7 Hz). |
| 86% |
|
7.5 cm in diameterIn a 500 mL four-necked flask equipped with two stirring blades,40 g of trityl olmesartan medoxomil,180 ml of acetic acid,40 g (1.0 volume) of water was added,The mixture was stirred at 40 C. for 2 hours to carry out deprotection reaction (reaction conversion: 98.4%Olmesartan medoxomil purity: 98.95%,Olmesartan dimer: 0.047%).Then,The reaction solution was cooled to 20 C.,80 g of water was added,After stirring at 20 C. for 1 hour,The precipitated triphenylmethanol was removed by vacuum filtration,To the obtained filtrate, 240 ml of 10% sodium hydrogen carbonate was added,The liquidity of the aqueous layer was adjusted to pH 3.5,After adding 400 ml of ethyl acetate and vigorously stirring,The aqueous layer was separated,An organic layer containing olmesartan medoxomil was obtained (olmesartan medoxomil purity: 99.35%Dimer: 0.011%).200 ml of ethyl acetate was distilled off from the organic layer,After stirring at 20 to 30 C. for 1 hour,The precipitated solid was collected by filtration under reduced pressure as a wet body.The obtained wet body was dried at 40 C. for 14 hours,24 g of crystals of olmesartan medoxomil were obtained (yield: 86%, olmesartan medoxomil purity: 99.71%, dimer: 0.010%). |
| 85% |
|
Example 1323.88 g of (V) were suspended in 92 ml of acetonitrile. The mixture was cooled to 5C in order to add 24.6 ml of 6N HCI. The mixture was stirred for 2 hours at 5C. 150 ml of toluene were added. The pH was adjusted to 5.7 with potassium bicarbonate. The mixture was filtered in order to eliminate part of the salts generated. The solid was washed with 30 ml of acetonitrile. The phases were separated. The acetonitrile present in the organic phase was distilled. The mixture was cooled and filtered. The solid was washed with toluene and water. (VI) was dried in a heat cabinet. 16.66 g of (VI) were obtained (85% yield). |
| 83.1% |
In methanol; ethyl acetate; at 45℃; for 8h; |
Take 40g of compound 5, Add 80mL of methanol, 330 mL of ethyl acetate, Warmed to 45 , After 8 hours of reaction, Add 860mL water, Insoluble matter was removed by filtration, The filtrate was concentrated, In oily liquid, Acetonitrile, Stirred under a solid precipitation, filter, Washed with ethanol, 23.2 g of white solid was obtained, The yield was 83.1% The HPLC purity was 99.2%. |
| 82% |
With acetic acid; In water; at 50℃; for 4h;Product distribution / selectivity; |
Example 2 Olmesartan medoxomil of formula I20 g of the starting compound of formula III were stirred up in 75 ml of acetic acid and after stirring for 10 minutes 30 ml of water were added dropwise during 5 minutes. Then, the suspension was put in a 50C bath and stirred for 4 hours. Then 16 ml of water were added dropwise in 5 minutes, the mixture was taken out of the bath and after 5 minutes it was put in a cooling bath with the temperature of 10C. After 20 minutes the separated insoluble fraction, containing the side product trityl alcohol, was aspirated and washed with a mixture of 4 ml of AcOH + 2 ml of water. 40 ml of acetone and then 70 ml of water were added to the filtrate at 30C, the separated product was aspirated and 1 1.5 g (82 %) of the product were obtained. |
| 82% |
With hydroxylamine hydrochloride; In ethanol; acetone; at 25 - 40℃; for 4h; |
21.6 g of triethylolmethane-containing methoxycinnamic acid (formula 1a), 3 g of hydroxylamine hydrochloride, 14 mL of acetone and 70 mL of ethanol were added to the reaction part, followed by stirring at 25 to 40 C for 4 hours. When the reaction was completed, the reaction solution was concentrated under reduced pressure. Ethyl acetate (80 mL) was added to the residue, and the mixture was stirred for 3 hours to precipitate crystals. The precipitated crystals were stirred at 20 to 25 C for 2 hours, filtered and washed with 20 mL of purified water. To the filtrate was added 40 mL of ethyl acetate, the temperature was cooled to 0 to 5 C, The pH of the filtrate was adjusted to pH 5.0 to 6.0 by adding 45 g of triethylamine to the filtrate, and crystals were slowly precipitated. The precipitated crystals were stirred at 0 to 5 C for 3 hours, filtered, washed with 30 mL of purified water and 30 mL of acetone, and dried under reduced pressure for 15 hours to obtain 12.3 g (yield 82%, purity 99.96%) of olmesartan methoxysilane (Formula 1b). |
| 76.81% |
With water; acetic acid; at 40 - 45℃; for 2h; |
Example 2: preparation of olmesartan medoxomil (I) as per the present invention: To a mixture of acetic acid ( 37.5 ml) and water (12.5 ml) was added trityl olmesartan medoxomil (III) (10 g) and heated at 40-45 C for 2 hours. Water (12.5 ml) was added and the reaction mixture was filtered to remove trityl alcohol. The filtrate was subjected to agitated thin film dryer, wherein the feed rate was of about 4 to 10 ml per minute, heating medium was jacketed hot water at 45-50 C and vacuum was 70-75 mm/Hg. Crude olmesartan medoxomil (I) was recovered form ATFD. HPLC purity: olmesartan medoxomil (I) (97.81 %); olmesartan acid impurity (II) (0.97%). To acetone (40 ml) crude olmesartan medoxomil (I) was added, the slurry was heated at 54-58 C for 30 minutes and then stirred for 1 -2 hours at 0-5 C, the solid was filtered. Wet solid was added to acetone (120 ml) and heated at 55-60C. The solution was filtered. From the filtrate about 95 ml of acetone was distilled out at atmospheric pressure. The concentrated mass was stirred at 25-30 C for 8-12 hours and then at 0-5 C for 1 -3 hours. The solid was filtered, washed with acetone and dried. Yield 5.3 g (76.81 %). HPLC purity: olmesartan medoxomil (I) (99.67 %); olmesartan acid impurity (II) (0.07%). |
| 57 - 78% |
With water; In acetonitrile; for 14h;Heating / reflux;Product distribution / selectivity; |
Example 5; Olmesartan medoxomil (V); Water (10 g) was added to a solution of the starting substance (III; 20 g) in acetonitrile (50 ml), and the mixture was heated to a mild boil for 14 h. Acetonitrile was evaporated, and, after dissolving in acetone (150 ml), the mixture was filtered through alumina and concentrated. After crystallization from the mixture tetrahydrofuran / ethyl acetate H g (78 %) of the product with an HPLC purity of 97.0 % was obtained. Recrystallization from methanol and water gave 1O g of the product with an HPLC purity of 99.3 %; m.p. 175- 177 0C.; Example 6; Olmesartan medoxomil (V); Water (10 g) was added to a solution of the starting substance (III; 10 g) in acetonitrile (50 ml), and the mixture was heated to a mild boil for 14 h. Acetonitrile was evaporated, and, after dissolving in acetone (150 ml), the mixture was filtered through silica gel and concentrated. After crystallization from acetonitrile, 4 g (57 %) of the product was obtained. After recrystallization from the mixture methyl tert-butyl ether / ethyl acetate, 3.4 g of the product with an HPLC purity of 99.4 % was obtained; m.p. 175-177 C. |
|
With water; In acetic acid; at 60℃; for 1.5h;pH 2.21 - 2.23;Product distribution / selectivity; |
A solution of MTT in 10 volumes of acetic acid (75%) was heated for 1.5 hrs at 60 C. until a pH of 2.21-2.23 was achieved, and the reaction was stirred until the amount of MTT was less than 2%. The mixture was evaporated to dryness. Ethyl acetate (EtOAc, 1 volume) was added to the residue and then evaporated again (twice). The resulting solid was dissolved in EtOAc (12 vol) and heated to reflux. The solution was cooled (2 C.) and stirred for 2 hrs. The product was filtered, washed (EtOAc, 1 vol), and dried on vacuum (45 C.). |
|
With water; In butan-1-ol; at 110℃; for 2.5h;pH 4.41;Product distribution / selectivity; |
A solution of MTT in an organic solvent and water (20%) was heated for 4-8 hrs at reflux. When the solvents were either acetonitrile (ACN), isopropyl alcohol (IPA) or t-butanol (t-BuOH), 1 volume of water was added, and the reaction was stirred until the amount of MTT was less than 2%. The mixture was evaporated to dryness. Ethyl acetate (EtOAc, 1 volume) was added to the residue and then evaporated again (twice). The resulting solid was dissolved in EtOAc (12 vol) and heated to reflux. The solution was cooled (2 C.) and stirred for 2 hrs. The product was filtered, washed (EtOAc, 1 vol), and dried on vacuum (45 C.). Table 1 shows the process details with different organic solvents: TABLE 1 Total solvent Time Solvent(s) Volume Temperature ( C.) (hrs) pH ACN:H2O 5:1 + 1 85 7 4.89-4.3 IPA:H2O 5:1 + 1 85 7 4.62-4.25t-BuOH:H2O 5:1 + 1 85 7 4.78-4.28n-propanol:H2O 5:1 reflux 2.5 4.3n-BuOH:H2O 5:1 110 2.5 4.412-BuOH:H2O 5:1 100 3 4.5iso-penthanol:H2O 5:1 100 3 5DMA:H2O 5:1 100 4 4.5DMF:H2O 5:1 100 4 4.5 |
|
With water; In isopropyl alcohol; at 85℃; for 7h;pH 4.25 - 4.62;Product distribution / selectivity; |
A solution of MTT in an organic solvent and water (20%) was heated for 4-8 hrs at reflux. When the solvents were either acetonitrile (ACN), isopropyl alcohol (IPA) or t-butanol (t-BuOH), 1 volume of water was added, and the reaction was stirred until the amount of MTT was less than 2%. The mixture was evaporated to dryness. Ethyl acetate (EtOAc, 1 volume) was added to the residue and then evaporated again (twice). The resulting solid was dissolved in EtOAc (12 vol) and heated to reflux. The solution was cooled (2 C.) and stirred for 2 hrs. The product was filtered, washed (EtOAc, 1 vol), and dried on vacuum (45 C.). Table 1 shows the process details with different organic solvents: TABLE 1 Total solvent Time Solvent(s) Volume Temperature ( C.) (hrs) pH ACN:H2O 5:1 + 1 85 7 4.89-4.3 IPA:H2O 5:1 + 1 85 7 4.62-4.25t-BuOH:H2O 5:1 + 1 85 7 4.78-4.28n-propanol:H2O 5:1 reflux 2.5 4.3n-BuOH:H2O 5:1 110 2.5 4.412-BuOH:H2O 5:1 100 3 4.5iso-penthanol:H2O 5:1 100 3 5DMA:H2O 5:1 100 4 4.5DMF:H2O 5:1 100 4 4.5 |
|
With water; In N,N-dimethyl-formamide; at 100℃; for 4h;pH 4.5;Product distribution / selectivity; |
A solution of MTT in an organic solvent and water (20%) was heated for 4-8 hrs at reflux. When the solvents were either acetonitrile (ACN), isopropyl alcohol (IPA) or t-butanol (t-BuOH), 1 volume of water was added, and the reaction was stirred until the amount of MTT was less than 2%. The mixture was evaporated to dryness. Ethyl acetate (EtOAc, 1 volume) was added to the residue and then evaporated again (twice). The resulting solid was dissolved in EtOAc (12 vol) and heated to reflux. The solution was cooled (2 C.) and stirred for 2 hrs. The product was filtered, washed (EtOAc, 1 vol), and dried on vacuum (45 C.). Table 1 shows the process details with different organic solvents: TABLE 1 Total solvent Time Solvent(s) Volume Temperature ( C.) (hrs) pH ACN:H2O 5:1 + 1 85 7 4.89-4.3 IPA:H2O 5:1 + 1 85 7 4.62-4.25t-BuOH:H2O 5:1 + 1 85 7 4.78-4.28n-propanol:H2O 5:1 reflux 2.5 4.3n-BuOH:H2O 5:1 110 2.5 4.412-BuOH:H2O 5:1 100 3 4.5iso-penthanol:H2O 5:1 100 3 5DMA:H2O 5:1 100 4 4.5DMF:H2O 5:1 100 4 4.5 |
|
With water; In dimethyl amine; at 100℃; for 4h;pH 4.5;Product distribution / selectivity; |
A solution of MTT in an organic solvent and water (20%) was heated for 4-8 hrs at reflux. When the solvents were either acetonitrile (ACN), isopropyl alcohol (IPA) or t-butanol (t-BuOH), 1 volume of water was added, and the reaction was stirred until the amount of MTT was less than 2%. The mixture was evaporated to dryness. Ethyl acetate (EtOAc, 1 volume) was added to the residue and then evaporated again (twice). The resulting solid was dissolved in EtOAc (12 vol) and heated to reflux. The solution was cooled (2 C.) and stirred for 2 hrs. The product was filtered, washed (EtOAc, 1 vol), and dried on vacuum (45 C.). Table 1 shows the process details with different organic solvents: TABLE 1 Total solvent Time Solvent(s) Volume Temperature ( C.) (hrs) pH ACN:H2O 5:1 + 1 85 7 4.89-4.3 IPA:H2O 5:1 + 1 85 7 4.62-4.25t-BuOH:H2O 5:1 + 1 85 7 4.78-4.28n-propanol:H2O 5:1 reflux 2.5 4.3n-BuOH:H2O 5:1 110 2.5 4.412-BuOH:H2O 5:1 100 3 4.5iso-penthanol:H2O 5:1 100 3 5DMA:H2O 5:1 100 4 4.5DMF:H2O 5:1 100 4 4.5 |
|
With water; In i-Amyl alcohol; at 100℃; for 3h;pH 5;Product distribution / selectivity; |
A solution of MTT in an organic solvent and water (20%) was heated for 4-8 hrs at reflux. When the solvents were either acetonitrile (ACN), isopropyl alcohol (IPA) or t-butanol (t-BuOH), 1 volume of water was added, and the reaction was stirred until the amount of MTT was less than 2%. The mixture was evaporated to dryness. Ethyl acetate (EtOAc, 1 volume) was added to the residue and then evaporated again (twice). The resulting solid was dissolved in EtOAc (12 vol) and heated to reflux. The solution was cooled (2 C.) and stirred for 2 hrs. The product was filtered, washed (EtOAc, 1 vol), and dried on vacuum (45 C.). Table 1 shows the process details with different organic solvents: TABLE 1 Total solvent Time Solvent(s) Volume Temperature ( C.) (hrs) pH ACN:H2O 5:1 + 1 85 7 4.89-4.3 IPA:H2O 5:1 + 1 85 7 4.62-4.25t-BuOH:H2O 5:1 + 1 85 7 4.78-4.28n-propanol:H2O 5:1 reflux 2.5 4.3n-BuOH:H2O 5:1 110 2.5 4.412-BuOH:H2O 5:1 100 3 4.5iso-penthanol:H2O 5:1 100 3 5DMA:H2O 5:1 100 4 4.5DMF:H2O 5:1 100 4 4.5 |
|
With water; In acetonitrile; at 85℃; for 7h;pH 4.3 - 4.89;Product distribution / selectivity; |
A solution of MTT in an organic solvent and water (20%) was heated for 4-8 hrs at reflux. When the solvents were either acetonitrile (ACN), isopropyl alcohol (IPA) or t-butanol (t-BuOH), 1 volume of water was added, and the reaction was stirred until the amount of MTT was less than 2%. The mixture was evaporated to dryness. Ethyl acetate (EtOAc, 1 volume) was added to the residue and then evaporated again (twice). The resulting solid was dissolved in EtOAc (12 vol) and heated to reflux. The solution was cooled (2 C.) and stirred for 2 hrs. The product was filtered, washed (EtOAc, 1 vol), and dried on vacuum (45 C.). Table 1 shows the process details with different organic solvents: TABLE 1 Total solvent Time Solvent(s) Volume Temperature ( C.) (hrs) pH ACN:H2O 5:1 + 1 85 7 4.89-4.3 IPA:H2O 5:1 + 1 85 7 4.62-4.25t-BuOH:H2O 5:1 + 1 85 7 4.78-4.28n-propanol:H2O 5:1 reflux 2.5 4.3n-BuOH:H2O 5:1 110 2.5 4.412-BuOH:H2O 5:1 100 3 4.5iso-penthanol:H2O 5:1 100 3 5DMA:H2O 5:1 100 4 4.5DMF:H2O 5:1 100 4 4.5 |
|
With water; In tert-butyl alcohol; at 85℃; for 7h;pH 4.28 - 4.78;Product distribution / selectivity; |
A solution of MTT in an organic solvent and water (20%) was heated for 4-8 hrs at reflux. When the solvents were either acetonitrile (ACN), isopropyl alcohol (IPA) or t-butanol (t-BuOH), 1 volume of water was added, and the reaction was stirred until the amount of MTT was less than 2%. The mixture was evaporated to dryness. Ethyl acetate (EtOAc, 1 volume) was added to the residue and then evaporated again (twice). The resulting solid was dissolved in EtOAc (12 vol) and heated to reflux. The solution was cooled (2 C.) and stirred for 2 hrs. The product was filtered, washed (EtOAc, 1 vol), and dried on vacuum (45 C.). Table 1 shows the process details with different organic solvents: TABLE 1 Total solvent Time Solvent(s) Volume Temperature ( C.) (hrs) pH ACN:H2O 5:1 + 1 85 7 4.89-4.3 IPA:H2O 5:1 + 1 85 7 4.62-4.25t-BuOH:H2O 5:1 + 1 85 7 4.78-4.28n-propanol:H2O 5:1 reflux 2.5 4.3n-BuOH:H2O 5:1 110 2.5 4.412-BuOH:H2O 5:1 100 3 4.5iso-penthanol:H2O 5:1 100 3 5DMA:H2O 5:1 100 4 4.5DMF:H2O 5:1 100 4 4.5 |
|
With water; In propan-1-ol; for 2.5h;pH 4.3;Heating / reflux;Product distribution / selectivity; |
A solution of MTT in an organic solvent and water (20%) was heated for 4-8 hrs at reflux. When the solvents were either acetonitrile (ACN), isopropyl alcohol (IPA) or t-butanol (t-BuOH), 1 volume of water was added, and the reaction was stirred until the amount of MTT was less than 2%. The mixture was evaporated to dryness. Ethyl acetate (EtOAc, 1 volume) was added to the residue and then evaporated again (twice). The resulting solid was dissolved in EtOAc (12 vol) and heated to reflux. The solution was cooled (2 C.) and stirred for 2 hrs. The product was filtered, washed (EtOAc, 1 vol), and dried on vacuum (45 C.). Table 1 shows the process details with different organic solvents: TABLE 1 Total solvent Time Solvent(s) Volume Temperature ( C.) (hrs) pH ACN:H2O 5:1 + 1 85 7 4.89-4.3 IPA:H2O 5:1 + 1 85 7 4.62-4.25t-BuOH:H2O 5:1 + 1 85 7 4.78-4.28n-propanol:H2O 5:1 reflux 2.5 4.3n-BuOH:H2O 5:1 110 2.5 4.412-BuOH:H2O 5:1 100 3 4.5iso-penthanol:H2O 5:1 100 3 5DMA:H2O 5:1 100 4 4.5DMF:H2O 5:1 100 4 4.5 |
|
With water; In iso-butanol; at 100℃; for 3h;pH 4.5;Product distribution / selectivity; |
A solution of MTT in an organic solvent and water (20%) was heated for 4-8 hrs at reflux. When the solvents were either acetonitrile (ACN), isopropyl alcohol (IPA) or t-butanol (t-BuOH), 1 volume of water was added, and the reaction was stirred until the amount of MTT was less than 2%. The mixture was evaporated to dryness. Ethyl acetate (EtOAc, 1 volume) was added to the residue and then evaporated again (twice). The resulting solid was dissolved in EtOAc (12 vol) and heated to reflux. The solution was cooled (2 C.) and stirred for 2 hrs. The product was filtered, washed (EtOAc, 1 vol), and dried on vacuum (45 C.). Table 1 shows the process details with different organic solvents: TABLE 1 Total solvent Time Solvent(s) Volume Temperature ( C.) (hrs) pH ACN:H2O 5:1 + 1 85 7 4.89-4.3 IPA:H2O 5:1 + 1 85 7 4.62-4.25t-BuOH:H2O 5:1 + 1 85 7 4.78-4.28n-propanol:H2O 5:1 reflux 2.5 4.3n-BuOH:H2O 5:1 110 2.5 4.412-BuOH:H2O 5:1 100 3 4.5iso-penthanol:H2O 5:1 100 3 5DMA:H2O 5:1 100 4 4.5DMF:H2O 5:1 100 4 4.5 |
|
With acetic acid; In water; toluene; |
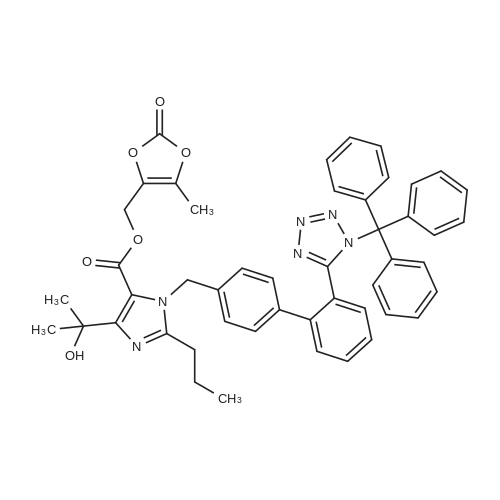
61(b) (5-Methyl-2-oxo-1,3-dioxolen-4yl)methyl 4-(1-hydroxy-1-methylethyl)-2-propyl-1-{4-[2-(tetrazol-5-yl)phenyl]phenyl}methylimidazole-5-carboxylate A mixture of 1.4 g of (5-methyl-2-oxo-1,3-dioxolen-4-yl)methyl 4-(1-hydroxy-1-methylethyl)-2-propyl-1-{4-[2-(trityltetrazol-5-yl)phenyl]phenyl}methylimidazole-5-carboxylate [prepared as described in step (a) above] and 48 ml of 75% v/v aqueous acetic acid was stirred at 60 C. for 1 hour, after which it was concentrated by evaporation under reduced pressure. The residue was dissolved in toluene, and the resulting solution was concentrated by distillation under reduced pressure; this was repeated a further time in order to remove the remaining water and acetic acid. The residue thus obtained was purified by column chromatography through silica gel using 1:9 and 1:4 by volume mixtures of methanol and methylene chloride as the eluent, to give 0.73 g of the title compound, melting at 170-172 C. Nuclear Magnetic Resonance Spectrum (CDCl3), delta ppm: 0.93 (3H, triplet, J=7.5 Hz); 1.63 (6H, singlet); 1.6-1.8 (2H, multiplet); 2.19 (3H, singlet); 2.70 (2H, triplet, J=7.5 Hz); 5.00 (2H, singlet); 5.45 (2H, singlet); 6.83 (2H, doublet, J=8 Hz); 7.10 (2H, doublet, J=8 Hz); 7.42-7.63 (3H, multiplet); 7.83 (1H, doublet of doublets, J=1 & 7.5 Hz). |
|
In water; acetic acid; toluene; |
78(b) (5-Methyl-2-oxo-1,3-dioxolen-4-yl)methyl 4-(1-hydroxy-1-methylethyl)-2-propyl-1-{4-[2-(tetrazol-5-yl)phenyl]phenyl}methylimidazole-5-carboxylate 75 ml of water were added to a suspension of 29.3 g of (5-methyl-2-oxo-1,3-dioxolen-4-yl)methyl 4-(1-hydroxy-1-methylethyl)-2-propyl-1-{4-[2-(trityltetrazol-5-yl)phenyl]phenyl}methylimidazole-5-carboxylate [prepared as described in step (a) above] in 225 ml of acetic acid, and the resulting mixture was stirred at 60 C. for 1.5 hours. At the end of this time, 75 ml of water were added to the mixture, which was then cooled. Precipitated trityl alcohol was removed by filtration, and the filtrate was concentrated by evaporation under reduced pressure. Toluene was added to the residue, and the mixture was again concentrated by evaporation under reduced pressure, to remove the remaining water and acetic acid. The residue was crystallized in ethyl acetate, to give 16.6 g of the title compound as crystals, melting at 177-180 C. (with decomposition). The Nuclear Magnetic Resonance Spectrum of this compound was identical with that of the compound obtained as described in Example 61(b). |
|
|
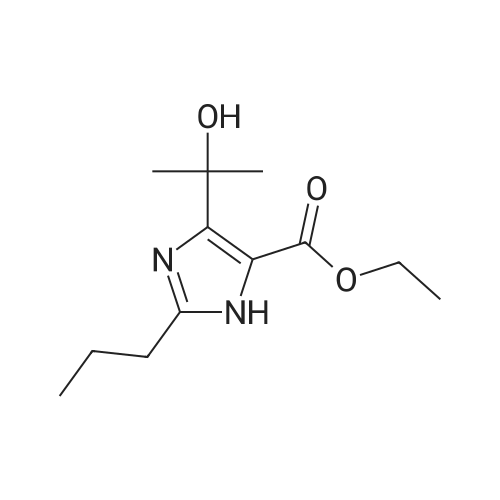
Example 1; Preparation of olmesartan medoxomilTo dimethyl acetamide (300 ml) was added 4-(1-hydroxy-1-methylethyl)-2-propyl imidazol- 5-carboxylic acid ethyl ester (50 gms) and powdered sodium hydroxide (26 gms). To this, 4-[2-(trityltetrazol-5-yl)phenyl]benzyl bromide (135 gms) was charged at 45-500C. The contents were stirred for 5 hours at 45-500C. Diisopropylethyl amine (100 ml) was charged to the reaction mass at 40-450C. A solution of 5-methyl-2-oxo-1 , 3-dioxane-4-yl)methyl chloride (80 gms) diluted with dimethyl acetamide (160 ml) was slowly added to the reaction mass at 40-450C over a period of 1 hour. The contents were heated to 60-650C and maintained for 4 hours. The reaction mass was then cooled to 30-350C and neutralized with concentrated hydrochloride acid. The reaction mass was filtered to remove inorganic impurities, charcoalized using charcoal (10 gms) andstirred for 30 minutes at 40-450C. The reaction mass was filtered over hyflo. The clear filtrate was acidified with hydrochloric acid (100 ml) slowly at 25-30C. The contents were stirred at 60C for 1 hour. The reaction mass was chilled to 0-5C and filtered to remove tritanol. The reaction mass was concentrated under reduced pressure. The residue was quenched with water (500ml), neutralized with base and extracted in dichloromethane (500 ml). The clear dichloromethane extract was then concentrated under reduced pressure and stripped off with acetone. The residue thus obtained was isolated from acetone (250 ml) to give 55 gms of the title compound. Chromatographic purity- > 99%; Example 2Preparation of olmesartan medoxomilTo dimethyl acetamide (600 ml) was added 4-(1-hydroxy-1-methylethyl)-2-propyl imidazol- 5-carboxylic acid ethyl ester (100 gms) and powdered potassium hydroxide (50 gms). To this was charged 4-[2-(trityltetrazol-5-yl)phenyl]benzyl bromide (270 gms) at 45-50C. The contents were stirred for 5 hours at 45-50C. Diisopropylethyl amine (200 ml) was charged to the reaction mass at 40-450C. To this was slowly added a solution of 5-methyl-2-oxo- 1 ,3-dioxane-4-yl)methyl chloride (160 gms) diluted with dimethyl acetamide (320 ml) at 40- <n="18"/>45C over a period of 1 hour. The contents were heated to 60-650C and maintained for 4 hours. The reaction mass was then cooled to 30-350C and was neutralized with concentrated hydrochloride acid. The reaction mass was filtered to remove inorganic impurities. The reaction mass was charcoalized using charcoal (20 gms) and was stirred for 30 minutes at 40-450C. The reaction mass was filtered over hyflo. The clear filtrate was acidified with hydrochloric acid (200 ml) slowly at 25-300C. The contents were stirred at 60C for 1 hour. The reaction mass was chilled to 0-50C and was filtered to remove tritanol. The reaction mass was concentrated under reduced pressure. The residue was quenched with water (1000 ml), neutralized with base and extracted in dichloromethane (1000 ml). The clear dichloromethane extract was then concentrated under reduced pressure, stripped off with acetone. The residue thus obtained was isolated from the acetone (500 ml) to give 110 gms of the title compound. Chromatogrphic purity- > 99%; Example 3Preparation of olmesartan medoxomilTo dimethyl acetamide (800 ml) was added 4-(1-hydroxy-1-methylethyl)-2-propyl imidazol- 5-carboxylic acid ethyl ester (100 gms) and powdered potassium carbonate (200 gms). To this was charged 4-[2-(trityltetrazol-5-yl)phenyl]benzyl bromide (300 gms) at 45-50C. The contents were stirred for 8-10 hours at 45-50C. The insolubles were filtered. The contents were cooled to 5-100C. Potassium tertiary butoxide (100 gms) was charged at a temperature below 45C. The reaction was maintained at 40-450C for 3 hrs. To this was slowly added 5-methyl-2-oxo-1 ,3-dioxane-4-yl) methyl chloride at 40-450C over a period of 1 hour. The contents were heated to 60-650C and maintained for 4 hours. The reaction mass was then cooled to 30-350C and was neutralized with concentrated hydrochloride acid. The reaction mass was filtered to remove inorganics. The reaction mass was charcoalized using charcoal (10 gms) and was stirred for 30 minutes at 40-450C. The reaction mass was filtered over hyflo. The clear filtrate was acidified with hydrochloric acid (100 ml) slowly at 25-30C. The contents were stirred at 60C for 1 hour. The reaction mass was chilled to 0-5C and was filtered to remove tritanol. The reaction mass was <n="19"/>concentrated under reduced pressure. The residue was quenched with water (500 ml), neutralized with base and extracted in dichloromethane (500 ml).The clear dichloromethane extract was then concentrated under reduced pressure, stripped off with acetone. The residue thus obtained was isolated from the acetone (250 ml) to give 55 gms of the title compound. Chromatogrphic purity- > 99% |
|
|
Example 7; Preparation of olmesartan medoxomilTo 75 % aqueous acetic acid (1000 ml) was slowly added trityl olmesartan medoxomil (110 gms)[prepared as described in example 5] at 25-30C. The contents were stirred at 600C for 1 hour. The reaction mass was chilled to 0-5C and filtered to remove tritanol. The reaction mass was concentrated under reduced pressure. The residue was quenched with water (500 ml), neutralized with a base and extracted in dichloromethane (500 ml). The clear dichloromethane extract was then concentrated under reduced pressure and stripped off with acetone. The residue thus obtained was isolated from the acetone (250 ml) to give 55 gms of the title compound. Chromatogrphic purity - > 99% |
|
|
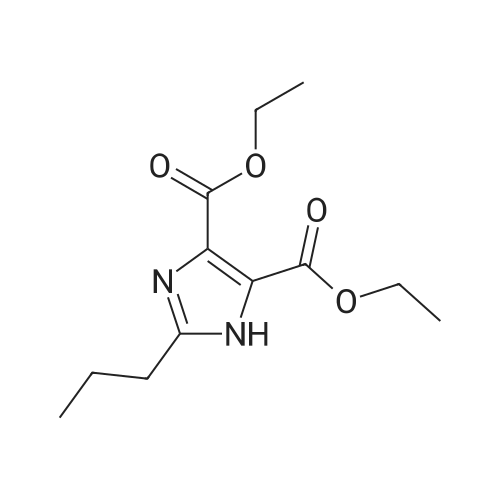
Example 236.0 g (50.3 mmol) ethyl 4-(1-hydroxy-1-methylethyl)-2-propyl-1-{4-[2-(trityltetrazol-5-yl)-phenyl]phenyl}-methyl imidazole-5-carboxylate (Va) and 3.0 g (75.4 mmol) of NaOH were suspended in 413 ml dimethylacetamide. The suspension was then stirred at room temperature for 20 h and after that 6.9 g (50.3 mmol) of K2CO3, were added. The mixture was cooled to 0 C. and solution of 15.4 g (70.4 mmol) 4-chloromethyl-5-methyl-2-oxo-1,3-dioxolene in 39 ml of dimethylacetamide were slowly added. The mixture was slowly heated to 50 C. and stirred at this temperature for 2 h. After esterification was completed, the mixture was cooled to 10 C. and poured into a mixture of 625 ml of ethyl acetate and 625 ml of 10% NaCl, and stirred at 25 C. for 15 min. The phases were separated and organic phase was washed 2× with 500 ml of 10% NaCl, dried over Na2SO4 and filtered. The filtrate was concentrated up to ? (approximately 270 g) at reduced pressure.To the resulting solution, 80 ml of ethanol and 8.3 ml (100 mmol) of conc. HCl were added and stirred at 24-26 C. for 3 h. To the cooled mixture 600 ml of water was added and pH of the suspension was estimated to 5 by addition of 5 M NaOH. The phases were stirred for 15 min and separated. Water phase was reextracted with 150 ml of ethyl acetate. Collected organic phases were dried over Na2SO4, filtered and concentrated under reduced pressure. 560 ml of ethyl acetate were added and the mixture was evaporated again. After that, 300 ml of ethyl acetate were added and the mixture was cooled to 20 C. and stirred for 1 h, filtered off and washed with 20 ml of fresh ethyl acetate. The yield of the product (I) was 21 g (75%). |
|
|
Trityl olmesartan medoxomil (250 g, 310 mmol) (97.3 % area) is dissolved in THF (1560 ml) and 48 % aqueous hydrobromic acid (70.6 ml, 625 mmol) is added slowly. The mixture is stirred for at 25 C. After 1 hour the precipitate forms. The mixture is stirred for 1 additional hour at 25 C, then cooled to -5 C and stirred for 1.5 hours at -5 C. The precipitate is filtered. 940 ml of THF is added to the precipitate and the mixture is stirred for 1 h at 25 C and then 1 hour at -5 C. Then precipitate is filtered off and washed with cold THF (150 ml). It is then dissolved in a mixture of water (875 ml) and acetone (440 ml). To a clear solution 5 % aqueous solution of NaHCO3 is added to raise pH to 5.15. The mixture is stirred for 1 hour at room temperature and 1 hour at 0 C. The precipitate is filtered, washed with water and then recrystallized from a mixture of acetonitrile (280 ml) and water (70 ml) to give 124.5 g of olmesartan medoxomil (99.68 % area) |
|
|
Example 2; 36.0 g (50.3 mmol) ethyl 4-(1-hydroxy-1-methylethyl)-2-propyl-1-(4-[2-(trityltetrazol-5-yl)-phenyl]phenyl}-methyl imidazole-5-carboxylate (Va) and 3.0 g (75.4 mmol) of NaOH were suspended in 413 ml dimethylacetamide. The suspension was then stirred at room temperature for 20 h and after that 6.9 g (50.3 mmol) of K2CO3 were added. The mixture was cooled to 0C and solution of 15.4 g (70.4 mmol) 4-chloromethyl-5-methyl-2-oxo-1,3-dioxolene in 39 ml of dimethylacetamide were slowly added. The mixture was slowly heated to 50C and stirred at this temperature for 2 h. After esterification was completed, the mixture was cooled to 10 C and poured into a mixture of 625 ml of ethyl acetate and 625 ml of 10 % NaCl, and stirred at 25 C for 15 min. The phases were separated and organic phase was washed 2x with 500 ml of 10 % NaCl, dried over Na2SO4 and filtered. The filtrate was concentrated up to ? (approximately 270 g) at reduced pressure. To the resulting solution, 80 ml of ethanol and 8.3 ml (100 mmol) of conc. HCl were added and stirred at 24-26C for 3h. To the cooled mixture 600 ml of water was added and pH of the suspension was estimated to 5 by addition of 5 M NaOH. The phases were stirred for 15 min and separated. Water phase was reextracted with 150 ml of ethyl acetate. Collected organic phases were dried over Na2SO4, filtered and concentrated under reduced pressure. 560 ml of ethyl acetate were added and the mixture was evaporated again. After that, 300 ml of ethyl acetate were added and the mixture was cooled to 20 C and stirred for 1h, filtered off and washed with 20 ml of fresh ethyl acetate. The yield of the product (I) was 21 g (75 %). |
|
|
A mixture of trityl olmesartan medoxomil (4.0 g), methanol (20 mL), and water (10 mL) is stirred at 25-35C for 15 minutes. Aqueous HCI (1 mL) is added drop-wise at 25-35C. The mixture is cooled to 0-50C. Water (30 mL) is added and the mixture is further stirred at 0-5C for 1 hour. The insoluble material is removed by filtration and the pH of the filtrate is adjusted to about 3-4 by adding 10% aqueous NaHCOs at 25-35C. The mixture is extracted with dichloromethane (3chi40 ml_). The organic layers are combined and washed with water (40 ml_). The solvent is distilled under reduced pressure below 400C. Ethyl acetate (10 ml_) is added to the residue and the mixture is heated to 50-600C and stirred for 1 hour. The mixture is cooled to 25-35C. The formed solid is filtered, washed with ethyl acetate (5 ml_), and dried under vacuum at 60-700C (yield 2.0 g). |
|
|
Example 7:Preparation of olmesartan medoxomil Trityl olmesartan medoxomil (260 gm) as obtained in example 1 was dissolved in toluene (2600 ml) and then added concentrated hydrochloric acid (156 ml) for 1 hour 30 minutes at room temperature. The reaction mass was maintained for 1 hour 30 minutes at room temperature and then added water (1000 ml). The reaction mass was stirred for 45 minutes at room temperature and the layers were separated. To the aqueous layer was added ethyl acetate (5000 ml) at room temperature. The reaction mass was cooled to 15 to 20C and pH of the reaction mass was adjusted to 4.5 to 5.5 with sodium carbonate (20%, 560 ml). The reaction mass was stirred for 20 minutes at 20C and the layers were separated. The organic layer was dried over sodium sulfate and ethyl acetate was distilled off completely under vacuum at below 45C to obtain a residual mass. To the residual mass was added ethyl acetate (400 ml) at 40C and then heated to 75 to 80C. The contents were maintained for 30 minutes at 75 to 80C. The reaction mass was cooled to room temperature and stirred for 1 hour. The reaction mass was further cooled to 10 to 15C and stirred for 1 hour 30 minutes, filtered. The solid obtained was dried at 40 to 45C for 4 hours to obtain 150 gm of olmesartan medoxomil.Olmesartan medoxomil: 98.6%;Olmesartan acid impurity: 0.32%;Trityl olmesartan medoxomil impurity: 0.35%Methyl olmesartan medoxomil impurity: 0.35%. |
| 36 g |
With sulfuric acid; In water; acetic acid; at 25 - 30℃; for 1h; |
To TOLM (75 g) were successively added acetic acid:water=1:1 (330 mL, 4.4 vol) and concentrated sulfuric acid (5.4 mL, 1.08 eq). The obtained mixture was stirred at 25C - 30C for 1 hr. Using TLC (thin layer chromatography) (TLC eluent: 10% methanol/methylene chloride, detection method: UV), complete disappearance of TOLM was confirmed. [0309] The reaction mixture was filtered and insoluble trityl alcohol was removed. The aqueous layer was adjusted to pH 2 - 3 by adding 25% aqueous sodium carbonate solution (initial pH of the reaction mixture was 4 - 4.5). The reaction mixture was stirred for 5 min, methylene chloride (225 mL, 3 vol) was added thereto, and the mixture was stirred for 5 min. Stirring was stopped, and the mixture was stood and partitioned. The aqueous layer was extracted with methylene chloride (2x225 mL, 2x3 vol), the extracts were combined with the organic layer, deionized water (375 mL, 5vol) was added, and the mixture was stirred for 5 min. Stirring was stopped, and the mixture was stood for 5 min and partitioned. To the organic layer was added saturated brine (375 mL, 5 vol), and the mixture was stirred for 5 min, left standing and partitioned. The organic layer was concentrated under reduced pressure at 40C - 45C to give crude OLM MDX (49 g, 93%) as a pale-yellow solid.To the crude OLM MDX (49 g, 1 eq) obtained in the above-mentioned (7) was added acetone (735 mL, 15 vol), and the mixture was stirred at 55C - 60C for 10 min. Furthermore, the reaction mixture was stirred at the same temperature for 15 min, and acetone was evaporated under normal pressure. Heating was stopped when a solid was precipitated, and the mixture was cooled to 25C - 30C. The precipitated solid was collected by filtration and dried with suction for 30 min to give OLM MDX (41 g, 83%). [0311] To the OLM MDX obtained above was added isopropyl alcohol (164 mL, 4 vol), and the mixture was heated to 55C - 60C, and stirred at 55C - 60C for 1 hr. Heating was stopped and the mixture was gradually cooled to 25C - 30C, and stirred at 25C - 30C for 30 min. The precipitated solid was filtered and dried with suction to give OLM MDX (41 g, 100%). [0312] The OLM MDX (41 g) obtained above and acetone (about 1 L) were heated to 55C - 60C, and stirred at 55C - 60C for 25 min. Acetone was evaporated under normal pressure until the mixture became cloudy, and the mixture was gradually cooled to 25C - 30C. The precipitated solid was collected by filtration and dried with suction for 30 min to give OLM MDX (34 g, 83%). The HPLC purity of the obtained OLM MDX was 99.66%. [0313] The OLM MDX (44 g) obtained above was dissolved in acetone (about 1.2 L), and the mixture was stirred at 55C-60C for 10 min. Acetone was evaporated under normal pressure until the solution became cloudy, and the solution was gradually cooled to 25C - 30C. The precipitated solid was collected by filtration, dried with suction for 30 min, blast dried for 1 hr, and further blast dried at 40C - 45C for 5 hr to give OLM MDX (36 g) as a white solid. The HPLC purity of the obtained OLM MDX was 99.8%. |
|
With acetic acid; In water; at 20 - 40℃; for 3h; |
To 2000mL four-neck flask equipped with two stirring bladesof diameter 15 cm, tritylolmesartan medoxomil100 g, acetic acid450 ml, water 150mLwere added, & stirred for 2 hours at 40 C and deprotection reaction was carried out. Thereaction solution was then cooled to 20 C, stirred for 1 hour at 20 C, and precipitated triphenyl methanol wasremoved by filtrationunder reduced pressure, and to the resulting filtrate, 10% sodium bicarbonate 500ml, ethyl acetate1000ml were added and after vigorousstirrind, the aqueous layer was separated, to obtain an organic layercontaining olmesartanmedoxomil. ethylacetate 400ml was evaporated from the organic layer, and stirred for 1 hour at20 ~ 30 , and the precipitated solid was done filtration under reduced pressure and fractionated aswet product. The resulting wet product was dried for 14 hours at 40 , the crude olmesartan medoxomil wasobtained 60g(residual ethyl acetate content: 5420ppm, the residual amount of acetic acid:490ppm, purity: 99.37%). In 100mL three-necked flask equipped with two stirringblades of diameter 2.5 cm, crude olmesartan medoxomil 5g obtained in ProductionExample 1, acetone 15 ml, ethyl acetate 15 mL, water 3g were added and heatedto 60 C, to obtain a solution of olmesartan medoxomil (solution adjustingstep). then cooled to 30 C, and after adding seed crystals of olmesartanmedoxomil, it was stirred for 15 hours at 28 C (crystallization step: it wasconfirmed that there is no change in the amount of crystals even if it isretained for more time.) . |
|
With acetic acid; at 50℃; for 2.5h;Cooling with ice; |
To 3L reaction flask 165.3g above prepared 1 - [[[2- (triphenylmethyl) -2H- tetrazole-5-yl] biphenyl-4-yl] methyl] -2- propyl-4- (1-hydroxy-1-methylethyl) imidazole-5-carboxylic acid (5-methyl-2-oxo-1,3-dioxolen-4-yl) methyl ester ( compound ) and 1.90L70% acetic acid, mechanical stirring, heated to 50 , the reaction 2.5h, an ice bath was added 1.1L of water was stirred.Filtration, the filtrate with 300ml * 3 times extracted with methylene chloride, the methylene chloride phase 150mL × 5 times with 5% acetic acid solution was washed with acetic acid solution and extracted with 250ml of dichloromethane, and the combined organic phases.The solvent was distilled off under reduced pressure, the residue was added 380mL of ethyl acetate was heated with stirring and cooling to precipitate a solid.Suction filtered, the filter cake was dried in vacuo to give a white solid product to 89.6 g, weight yield was 77.8%.To a 1L reaction flask above 85g crude product olmesartan medoxomil, 420ml of tetrahydrofuran was heated at reflux, was added 360ml of ethyl acetate, the ice bath was stirred for crystallization.Filtration cake blast drying to constant weight to give the product as a white solid 74.8g, a yield of 83.5% by weight. |
|
With sulfuric acid; In water; acetone; at 30℃; for 4h; |
(4) the C48 H44 N6 O6 AMST - 6, acetone, water, sulfuric acid added to the reactor, the temperature rising to 30 C reaction 2.5 hours, adding water, 30 C continue to reaction 1.5 hours or more, when the raw material to achwhich isve the requirement of lowering the temperature to 5 C stirring 1 or more hours, filtering the triphenyl methanol, the filter cake is washed with water; mother liquor up to 50 C, add sodium bicarbonate, stirring 1 hour, filtration products, the filter cake is washed with water, 50 C decompression drying 12 hours or more to constant weight, to obtain omay sha tanzhi thick; said C48 H44 N6 O6 AMST - 6, acetone, water, sulfuric acid and sodium bicarbonate and the mass ratio of 700:1400:: 2101.4: 194.7: 373.5; (5) the crude product of olmesartan medoxomil and acetone is added to the reaction in the bottle, heat to reflux to totally dissolve, filter press, distilling off acetone, lowering the temperature to -10 C crystallization, filtration, acetone washing, 50 C drying productC29 H30 N6 O6 Olmesartan medoxomil, the crude with olmesartan medoxomil mass ratio of olmesartan medoxomil 448.4: 6500; |
| 44 g |
With acetic acid; at 45 - 55℃; for 2h;Green chemistry; |
1L glass reaction flask was added with 500ml of 75% acetic acid, 100.0g of intermediate 2 was added with stirring, and the mixture was heated to stirAfter 45-55 C, after 2 hours of incubation, reduce the temperature to 20-30 C, add 125ml of water, stir for 30 minutes, filter, filter cake with 100ml of 75% acetic acid, transfer the filtrate to 3L glass reaction bottle, The filtrate was successively added with 1000 ml of dichloromethane and 1000 ml of water, and the mixture was stirred and the organic layer was temporarily stored. The aqueous layer was extracted with 500 ml of dichloromethane, and the organic layer was combined. The organic layer was washed with 1000 ml of water, and the organic layer was washed with 5% carbonic acid. The aqueous sodium hydrogen solution was adjusted to rhoEta=6.0~7.0 (about 600 ml), and the organic layer was washed with 500 ml of water, and the organic layer was separated. The organic layer was concentrated under reduced pressure at 30-40 C until no significant solvent was discharged, and 300 ml was added. Ethyl acetate was stirred and crystallized at 20-30 C for 1 hour, filtered, and the filter cake was rinsed with 300 ml of ethyl acetate. The filter cake was blast dried at 40-50 C for 8 hours to obtain a white solid 60.0 g. (product), yield 86.0%, HPLC: 99.0%. |
| 30 g |
With acetic acid; In water; at 20 - 40℃; for 3h; |
Into a 1000 mL four-necked flask equipped with a two-piece stirring blade having a diameter of 10 cm,50 g of trityl olmesartan medoxomil, 225 ml of acetic acid and 75 ml of water were added and stirred at 40 C. for 2 hours to carry out deprotection reaction.Subsequently, the reaction solution was cooled to 20 C. and stirred at 20 C. for 1 hour, and the precipitated triphenylmethanol was removed by vacuum filtration,To the obtained filtrate were added 250 ml of 10% sodium hydrogen carbonate and 500 ml of ethyl acetate, followed by vigorous stirring, and the aqueous layer was separated to obtain an organic layer containing olmesartan medoxomil.200 ml of ethyl acetate was distilled off from the organic layer, and the mixture was stirred at 20 to 30 C. for 1 hour. The precipitated solid was collected by filtration under reduced pressure as a wet body.The obtained wet body was dried at 40 C. for 14 hours to obtain 30 g of crystals of olmesartan medexomil (purity: 99.54%). |






 Chemistry
Chemistry
 Pharmaceutical Intermediates
Pharmaceutical Intermediates
 Inhibitors/Agonists
Inhibitors/Agonists
 Material Science
Material Science







 For Research Only
For Research Only
 120K+ Compounds
120K+ Compounds
 Competitive Price
Competitive Price
 1-2 Day Shipping
1-2 Day Shipping