| 27% |
With sodium tetrahydroborate; nickel dichloride; In ethanol; at 0 - 20℃; for 3h; |
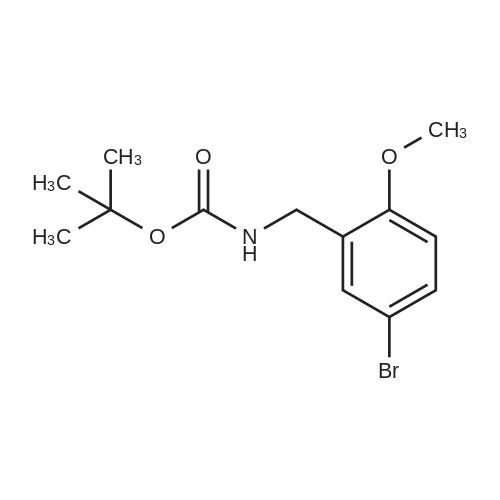
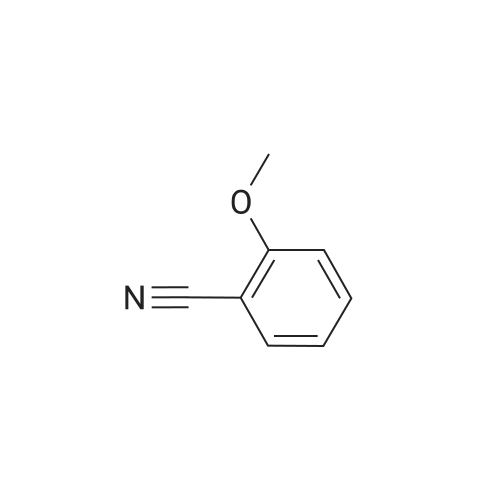
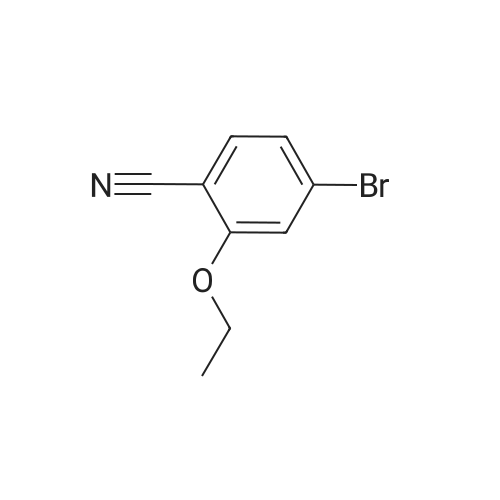
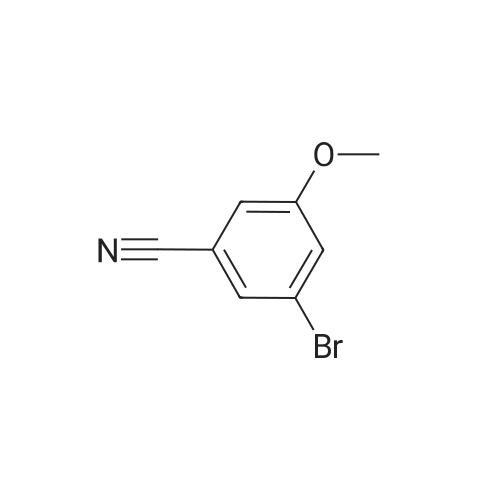
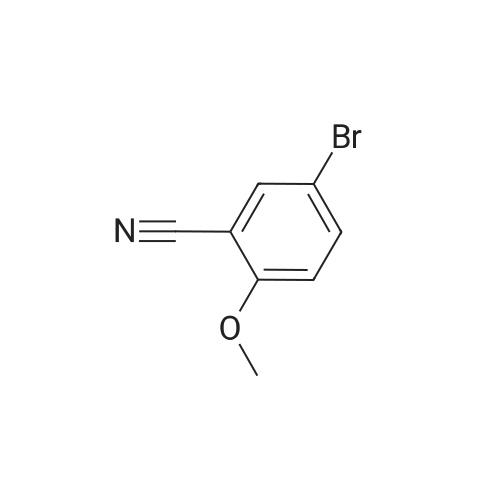
Example 38 1,1-Dimethylethyl [5-bromo-2-(methyloxy)phenyl]methyl}carbamate NaBH4 (2.9 g, 75.5 mmol) was cautiously added in several portions to a solution of NiCl2 (2.6 g, 19.8 mmol), Boc2O (8.2 g, 37.7 mmol) and <strong>[144649-99-0]5-bromo-2-(methyloxy)benzonitrile</strong> (4.0 g, 18.9 mmol) in dry EtOH (70 mL) at 0° C. Once the reaction had subsided, the mixture was left to stir at room temperature for 3 h. Ethanol was removed under reduced pressure and the residue was dissolved in EtOAc and saturated solution of NaHCO3, then filtered and repeatedly washed with EtOAc. The combined organic phases were dried using Na2SO4. The crude product was purified by flash column chromatography (PE/EA=20:1) to give 1.6 g of the product, 1,1-dimethylethyl [5-bromo-2-(methyloxy)phenyl]methyl}-carbamate. (Yield: 27percent). 1H NMR (400 MHz, CDCl3): delta 7.36-7.33 (m, 2H), 6.74 (d, J=8.8 Hz, 1H), 4.97 (br, 1H), 4.27 (d, J=4.8 Hz, 1H), 3.82 (s, 3H), 1.45 (s, 9H); 13C NMR (400 MHz, CDCl3): delta 156.5, 155.8, 131.7, 131.1, 129.3, 111.8, 79.5, 55.5, 39.9, 26.4. HPLC: retention time: 6.592 min; purity: 97.9percent. |
| 25% |
With sodium tetrahydroborate; ethanol;nickel dichloride; at 0 - 20℃; |
Example 35 1,1-Dimethylethyl[5-bromo-2-(methyloxy)phenyl]methyl}carbamate NaBH4 (2.9 g, 75.5 mmol) was cautiously added in several portions to a solution of NiCl2 (2.6 g, 19.8 mmol), Boc2O (8.2 g, 37.7 mmol) and <strong>[144649-99-0]5-bromo-2-(methyloxy)benzonitrile</strong> (4.0 g, 18.9 mmol) in dry EtOH (70 mL) at 0° C. Once the reaction had subsided, the mixture was left to stir at room temperature for 3 h. Ethanol was removed under reduced pressure and the residue was dissolved in EtOAc and saturated solution of NaHCO3, then filtered and the aqueous layer was repeatedly washed with EtOAc. The combined organic phases were dried Na2SO4. The crude product was purified by flash column chromatography to give the captioned the product (1.5 g yield: 25percent). 1H NMR (400 MHz, CDCl3) delta 7.36-7.33 (m, 2H), 6.74 (d, J=8.8 Hz, 1H), 4.97 (br, 1H), 4.27 (d, J=4.8 Hz, 1H), 3.82 (s, 3H), 1.45 (s, 9H); 13C NMR (400 MHz, CDCl3) delta 156.5, 155.8, 131.7, 131.1, 129.3, 111.8, 79.5, 55.5, 39.9, 26.4. HPLC: retention time. |
| 25% |
With sodium tetrahydroborate; ethanol; nickel dichloride; at 0 - 20℃; for 3h; |
Example 35 1,1-Dimethylethyl [5-bromo-2-(methyloxy)phenyl] methyl} carbamateNaBH4 (2.9 g, 75.5 mmol) was cautiously added in several portions to a solution OfNiCl2 (2.6 g, 19.8 mmol), BoC2O (8.2 g, 37.7 mmol) and <strong>[144649-99-0]5-bromo-2-(methyloxy)benzonitrile</strong> (4.0 g, 18.9 mmol) in dry EtOH (70 mL) at 0 0C. Once the reaction had subsided, the mixture was left to stir at room temperature for 3 h. Ethanol was removed under reduced pressure and the residue was dissolved in EtOAc and saturated solution of NaHCpsi3, then filtered and the aqueous layer was repeatedly washed with EtOAc. The combined organic phases were dried Na2SO4. The crude product was purified by flash column chromatography to give the captioned the product (1.5g yield: 25percent). 1H NMR (400 MHz, CDCl3) delta 7.36-7.33 (m, 2 H), 6.74 (d, J=8.8 Hz, 1 H), 4.97 (br, 1 H), 4.27(d, J=4.8 Hz, 1 H), 3.82 (s, 3 H), 1.45 (s, 9 H); 13C NMR (400 MHz, CDCl3) delta 156.5, 155.8, 131.7, 131.1, 129.3, 111.8, 79.5, 55.5, 39.9, 26.4. HPLC: retention time. |






 Chemistry
Chemistry
 Pharmaceutical Intermediates
Pharmaceutical Intermediates
 Inhibitors/Agonists
Inhibitors/Agonists
 Material Science
Material Science







 For Research Only
For Research Only
 120K+ Compounds
120K+ Compounds
 Competitive Price
Competitive Price
 1-2 Day Shipping
1-2 Day Shipping