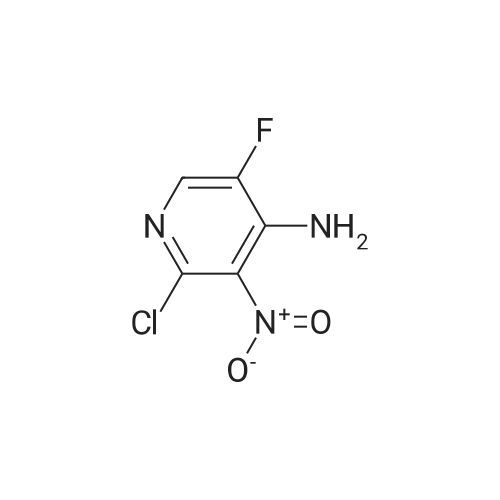
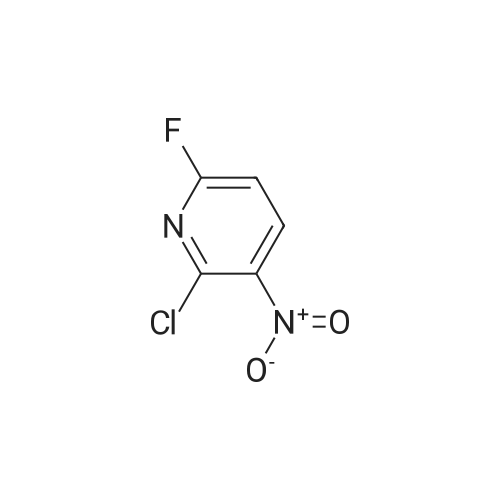
* All experimental methods are cited from the reference, please refer to the original source for details. We do not guarantee the accuracy of the content in the reference.
With benzyltrimethylammonium chloride; trichlorophosphate; In acetonitrile; at 80℃; for 6h;
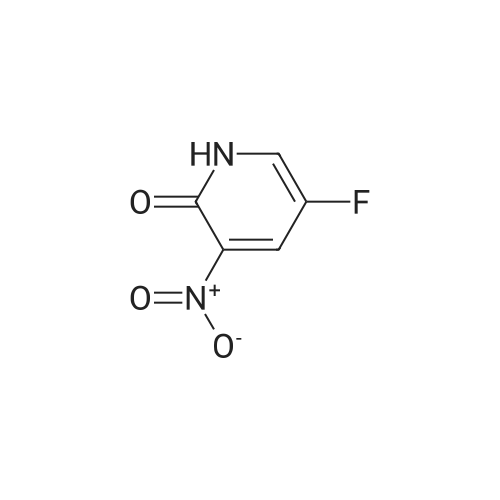
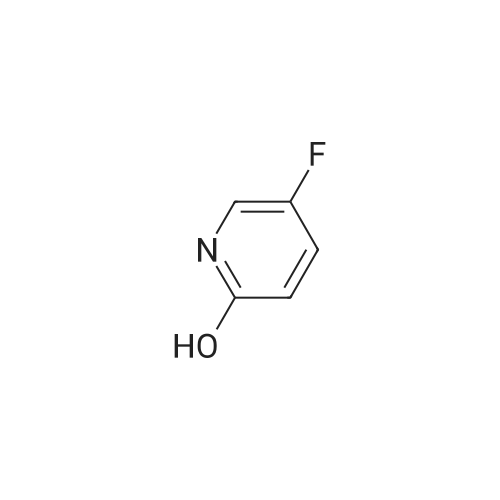
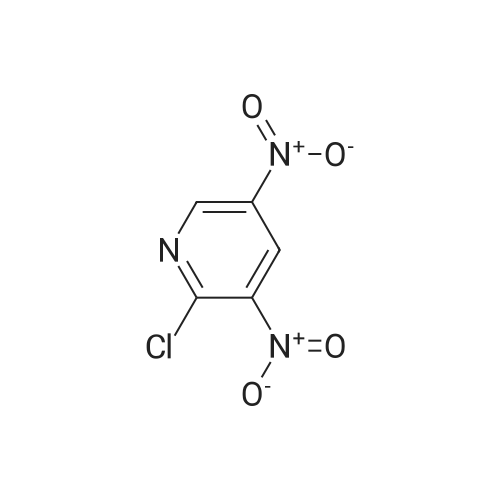
a) Synthesis of 2-chloro-5-fluoro-3-nitro-pyridine 5-fluoro-3-nitro-pyridin-2-ol (2 g, 12.7 mmol) and benzyltrimethyl ammonium chloride (1.17 g, 6.35 mmol) were dissolved in acetonitrile, and phosphorus oxychloride (3.5 ml, 38.1 mmol) was added thereto and stirred at 80 C. for 6 hours. The reaction mixture was cooled and poured into ice water to quench the reaction, and extracted with dichloromethane. The combined organic layer was washed with saturated saline solution, dried over anhydrous sodium sulfate (Na2SO4), filtered and evaporated under reduced pressure. The residue was purified by column chromatography on silica eluding with a solvent of dichloromethane:methanol=30:1. The fractions containing the product were collected and evaporated to obtain yellow liquid (1.57 g, 70%). 1H-NMR (CDCl3, 300 MHz); delta=8.56 (d, J=2.7 Hz, 1H), 8.40 (dd, J=6.5 Hz, 2.7 Hz, 1H). MS (ESI); 176.9 (M++1).
69%
With tetraethylammonium chloride; trichlorophosphate; In acetonitrile; at 90℃; for 24h;
PREPARATION 53 2-Chloro-5-fluoro-3-nitropyridine 5-Fluoro-3-nitro-pyridin-2-ol (1.00 g, 5.33 mmol) was dissolved in acetonitrile (40 mL) and tetraethylammonium chloride (2.10g, 12.65 mmol) was added. The mixture became clear, phosphorous oxytrichloride (1.94 g, 12.65 mmol) was added at room temperature and the mixture was heated at 90 oC for 24h. The reaction mixture was evaporated to dryness; the residue was taken up with water (100 mL) and extracted with ethyl acetate (2x100 mL). The organic layer was dried with sodium sulfate, filtered and evaporated to dryness. A yellow solid (0.90 g, 69%) was isolated, pure enough to perform the next synthetic step. LRMS (m/z): 177 (M+1)+
41%
With trichlorophosphate;N,N-dimethyl-formamide; at 110℃; for 18h;
A mixture of the pyridine of preparation 55 (105 mg, 0.66 mmol), phosphorus oxychloride (1 ml) and Lambda/,Lambda/-dimethyIformamide (10 mul, catalytic) was heated at 11O0C for 18 hours. The cooled reaction mixture was then concentrated under reduced pressure. The crude product was purified by column chromatography on silica gel using dichloromethane/acetonitrile as eluant (100:0 to 50:50 v/v) to yield the title compound (48 mg, 41%) as a solid. 1H NMR (400MHz, CDCI3): delta 8.03 (dd, 1 H), 8.55 (d, 1 H).
With trichlorophosphate; In 1,2-dimethoxyethane; at 110℃; for 5h;
In Step B, compound zz2' (500 mg, 3.16 mmol) was dissolved in phosphorous oxychloride (1.7 mL, 18.9 mmol) and dimethoxyethane at room temperature. The reaction was heated to 110 C. for 5 hours. The excess phosphorous oxychloride was then removed by concentrating the reaction mixture in vacuo. The residue was chromatographed on silica gel, eluted with chloroform (100%) to afford 176 mg of product zz3'.
With tetraethylammonium chloride; trichlorophosphate; In acetonitrile; at 20 - 90℃; for 24h;
5-Fluoro-3-nitro-pyridin-2-ol (1 .00 g, 5.33 mmol) was dissolved in acetonitrile (40 mL) and tetraethylammonium chloride (2.1 Og, 12.65 mmol) was added. The mixture became clear, phosphorous oxytrichloride (1 .94 g, 12.65 mmol) was added at room temperature and the mixture was heated at 90 C for 24h. The reaction mixture was evaporated to dryness; the residue was taken up with water (100 mL) and extracted with ethyl acetate (2x100 mL). The organic layer was dried with sodium sulphate, filtered and evaporated to dryness. A yellow solid (0.90 g, 69%) was isolated, pure enough to perform the next synthetic step.LRMS (m/z): 177 (M+1 )+
With hydrogenchloride; In dimethylformamide [DMF]; ethyl acetate; N,N-dimethyl-formamide;
C. 6-Fluoro-4-azaoxindole In a dry flask was placed sodium hydride as a 60% suspension in oil (3.1 g, 77.5 mmol). Most of the oil was removed by washing twice with hexanes. The remaining solid sodium hydride was then suspended in dry dimethylformamide (DMF) (100 mL) and cooled to 0 C. Diethyl malonate (11.8 mL, 77.7 mmol) was then added dropwise with stirring. The mixture was stirred at 0 C. for 1 hour after which a solution of <strong>[136888-21-6]2-chloro-5-fluoro-3-nitropyridine</strong> (5.21 g, 29.5 mmol) in DMF (40 mL) was added. Stirring at room temperature was continued overnight and the reaction mixture was poured into ice/water. Following acidification to pH 3 with 6N HCl solution, the mixture was extracted with ethyl acetate. The organic phase was washed with brine, dried over MgSO4 and concentrated in vacuo to leave a red oil. This was subjected to flash chromatography on silica gel using 3:7 ethyl acetate/hexanes, as eluant. All fractions containing the desired product were combined and concentrated to yield an oil containing 2-bis(ethoxycarbonyl)methyl-5-fluoro-3-nitropyridine and diethyl malonate in a molar ratio of approximately 11:9 weighing 11.5 g. The yield of 2-bis(ethoxycarbonyl)methyl-5-fluoro-3-nitropyridine was calculated to be approximately 8 g (90%).
With hydrogenchloride; In dimethylformamide [DMF]; ethyl acetate; N,N-dimethyl-formamide;
C. 6-Fluoro-4-azaoxindole In a dry flask was placed sodium hydride as a 60% suspension in oil (3.1 g, 77.5 mmol). Most of the oil was removed by washing twice with hexanes. The remaining solid sodium hydride was then suspended in dry dimethylformamide (DMF) (100 mL) and cooled to 0C. Diethyl malonate (11.8 mL, 77.7 mmol) was then added dropwise with stirring. The mixture was stirred at 0C for 1 hour after which a solution of <strong>[136888-21-6]2-chloro-5-fluoro-3-nitropyridine</strong> (5.21 g, 29.5 mmol) in DMF (40 mL) was added. Stirring at room temperature was continued overnight and the reaction mixture was poured into ice/water. Following acidification to pH 3 with 6N HCl solution, the mixture was extracted with ethyl acetate. The organic phase was washed with brine, dried over MgSO4 and concentrated in vacuo to leave a red oil. This was subjected to flash chromatography on silica gel using 3:7 ethyl acetate/hexanes, as eluant. All fractions containing the desired product were combined and concentrated to yield an oil containing 2-bis(ethoxycarbonyl)methyl-5-fluoro-3-nitropyridine and diethyl malonate in a molar ratio of approximately 11:9 weighing 11.5 g. The yield of 2-bis(ethoxycarbonyl)methyl-5-fluoro-3-nitropyridine was calculated to be approxiamtely 8 g (90%).
With sodium hydride; In N,N-dimethyl-formamide; at 20℃;Cooling with ice;
Diethyl malonate (9.98 g, 62.3 mmol) was diluted in DMF (50 ml), to which was then added 60 wt%sodium hydride (2.266 g, 56.6 mmol) under ice cooling, and the resulting mixture was stirred under ice cooling and at room temperature. After that, a solution of Compound 33 (5.00 g, 28.3 mmol) dissolved in DMF (5 ml) was added to the reaction mixture under ice cooling, and the resulting mixture was stirred at room temperature. After completion of the reaction, ethyl acetate was added to the reaction mixture under ice cooling, and the resulting mixture was washed with 2 mol/L aqueous solution of hydrochloric acid and with water. The obtained organic layer was dried over magnesium sulfate, and then the solvent was removed by concentration under reduced pressure. The obtained residue was purified by silica gel column chromatography. About 80% portion of the purified product was diluted in DMSO (50 ml), to which were then added water (0.5 ml) and lithium chloride (4.86 g, 115 mmol), and the resulting mixture was stirred at 110C. After completion of the reaction, a mixed solvent of ethyl acetate and hexane was added to the reaction mixture, from which insoluble materials were removed, and which was then washed with water. The obtained organic layer was dried over magnesium sulfate, and then the solvent was removed by concentration under reduced pressure. The obtained residue was purified by silica gel column chromatography to obtain Compound 122 (2.98 g). Compound 122; Method B LC/MS retention time = 1.65 min. MS (ESI) m/z = 229.15(M+H)+.
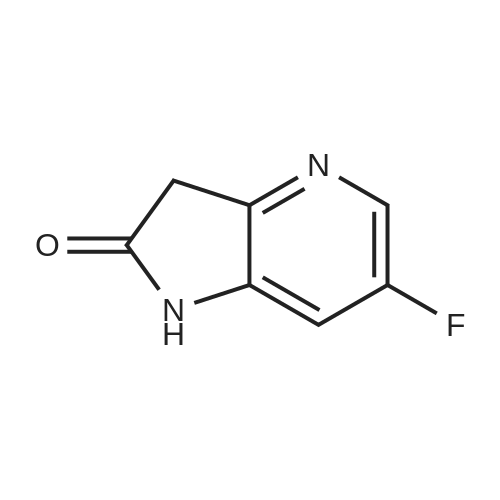
7-chloro-4-fluoro-1H-pyrrolo[2,3-c]pyridine[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
In tetrahydrofuran; at -20℃; for 15h;
In Step C, compound zz3' (140 mg, 0.79 mmol) was dissolved in THF (5 mL) and cooled to -78 C. under a nitrogen atmosphere. To this solution was added dropwise a solution of vinyl magnesium bromide (1.2 mmol, 1.0 M in diethyl ether, 1.2 mL). The reaction mixture was then kept at -20 C. for 15 hours. The reaction mixture was then quenched with saturated ammonium chloride, and extracted with ethyl acetate. The combined organic layers were washed with brine, dried over magnesium sulfate, filtered, and the filtrate was concentrated in vacuo. The residue was chromatographed on silica to provide 130 mg of precursor 2d 1H NMR (500 MHz, CD3OD) delta7.78 (s, 1H), 7.60 (d, 1H, J=3.0 Hz), 6.71 (d, 1H, J=3.05 Hz). MS m/z: (M+H)+ calcd for C7H5ClFN2: 171.10; found 171.00. HPLC retention time: 1.22 minutes (column A).
B. 2-Chloro-5-fluoro-3-nitropyridine To a mixture of phosphorus pentachloride (9.41 g, 45.2 mmol) and phosphorus oxychloride (4.2 mL, 45.1 mmol) at 60 C. was added in portions 5-fluro-2-hydroxy-3-nitropyridine (6.5 g, 41.1 mmol). The mixture was stirred in an oil bath at 100 C. under nitrogen overnight, cooled to room temperature and poured into ice/water. After addition of more water and ethyl acetate, the mixture was filtered through Celite to remove dark insoluble material. The organic phase was washed with brine, filtered again to remove more dark material, dried over MgSO4 and concentrated. The residue was subjected to flash chromatography on silica gel using chlorform as eluant. Fractions containing the title compound were combined and concentrated to provide a yellow oil (3.51 g, 48%) which solidified on standing at 5 C. overnight.
48%
B. 2-Chloro-5-fluoro-3-nitropyridine To a mixture of phosphorus pentachloride (9.41 g, 45.2 mmol) and phosphorus oxychloride (4.2 mL, 45.1 mmol) at 60C was added in portions 5-fluro-2-hydroxy-3-nitropyridine (6.5 g, 41.1 mmol). The mixture was stirred in an oil bath at 100C under nitrogen overnight, cooled to room temperature and poured into ice/water. After addition of more water and ethyl acetate, the mixture was filtered through CeliteRto remove dark insoluble material. The organic phase was washed with brine, filtered again to remove more dark material, dried over MgSO4 and concentrated. The residue was subjected to flash chromatography on silica gel using chlorform as eluant. Fractions containing the title compound were combined and concentrated to provide a yellow oil (3.51 g, 48%) which solidified on standing at 5C overnight.
9
1-[4-(4-chlorophenyl)-5-methyl-4H-1,2,4-triazol-3-yl]piperidin-4-amine dihydrochloride[ No CAS ]
[ 136888-21-6 ]
[ 913264-51-4 ]
Yield
Reaction Conditions
Operation in experiment
80%
The amine of preparation 49 (166 mg, 0.464 mmol) was dissolved in dichloromethane (10 ml) and the solution was treated with 1 N sodium hydroxide solution (10 ml). The layers were separated, and the organic phase was dried over magnesium sulfate, filtered and evaporated under reduced pressure. The pyridine of preparation 56 (41 mg, 0.23 mmol) was added to a solution of this amine in tetrahydrofuran (3 ml). The reaction mixture was heated at reflux for 18 hours. The cooled mixture was then concentrated under reduced pressure. The crude product was purified by column chromatography on silica gel using dichloromethane/methanol/ammonia as eluant (99:1 :0 to 95:5:0.5 v/v/v) to yield the title compound (80 mg, 80%) as a solid.1H NMR (400MHz, CD3OD): delta 1.52-1.62 (m, 2H), 1.96-2.02 (m, 2H), 2.23 (s, 3H), 2.95 (t, 2H), 3.29- 3.33 (m, 2H), 4.28 (m, 1 H), 7.51 (d, 2H), 7.64 (d, 2H), 8.29 (dd, 1 H), 8.42 (s, 1 H); LRMS APCI+ m/z 432 [MH]+.
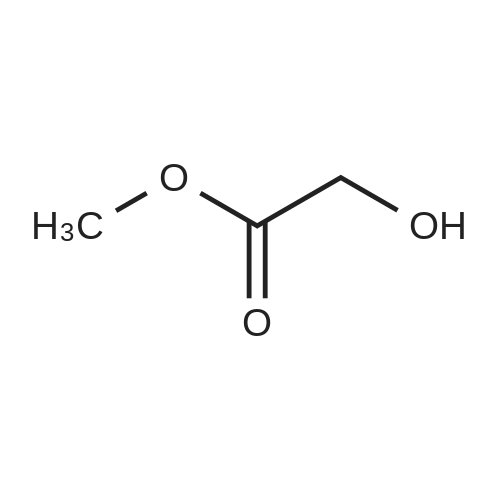
b) Synthesis of (5-fluoro-3-nitro-pyridin-2-yloxy)-acetic acid methyl ester Hydroxy-acetic acid methyl ester (0.96 g, 10.7 mmol) was dissolved in 2 ml of anhydrous tetrahydrofuran under nitrogen atmosphere, and sodium hydride (0.42 g, 10.7 mmol) was added thereto at room temperature. After stirring for 30 minutes, <strong>[136888-21-6]2-chloro-5-fluoro-3-nitro-pyridine</strong> (1.57 g, 8.89 mmol) dissolved in 15 ml of anhydrous tetrahydrofuran was added dropwise at room temperature, and the reaction mixture was stirred at room temperature for 18 hours. After completion of the reaction by adding water, the mixture was extracted with dichloromethane, and the combined organic layer was washed with water and saturated saline solution, dried over anhydrous sodium sulfate (Na2SO4), filtered and evaporated under reduced pressure. The residue was purified by column chromatography on silica eluding with a solvent of n-hexane:ethyl acetate=9:1. The fractions containing the product were collected and evaporated to obtain (5-fluoro-3-nitro-pyridin-2-yloxy)-acetic acid methyl ester as yellow liquid (1.35 g, 66%). 1H-NMR (CDCl3, 300 MHz); delta=8.25 (d, J=2.7 Hz, 1H), 8.40 (dd, J=6.9 Hz, 2.7H, 1 Hz), 5.06 (s, 2H), 3.78 (s, 3H). MS (ESI); 231.1 (M++1).






 Chemistry
Chemistry
 Pharmaceutical Intermediates
Pharmaceutical Intermediates
 Inhibitors/Agonists
Inhibitors/Agonists
 Material Science
Material Science







 For Research Only
For Research Only
 120K+ Compounds
120K+ Compounds
 Competitive Price
Competitive Price
 1-2 Day Shipping
1-2 Day Shipping