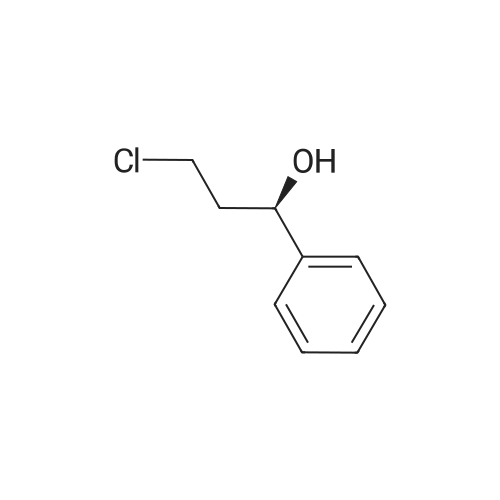
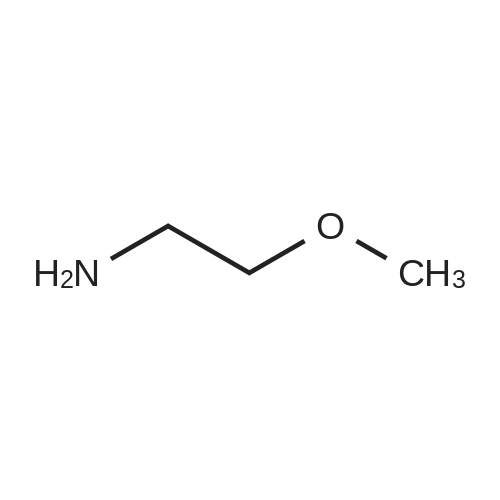
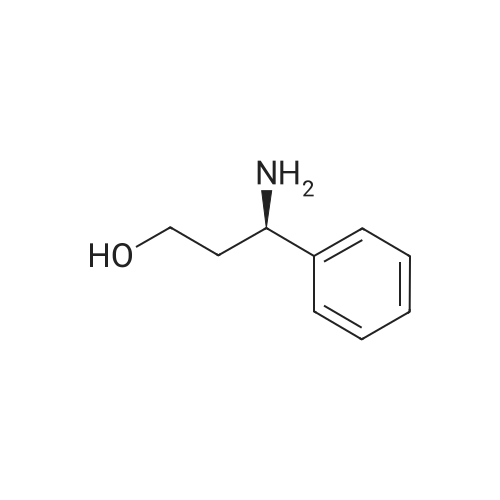
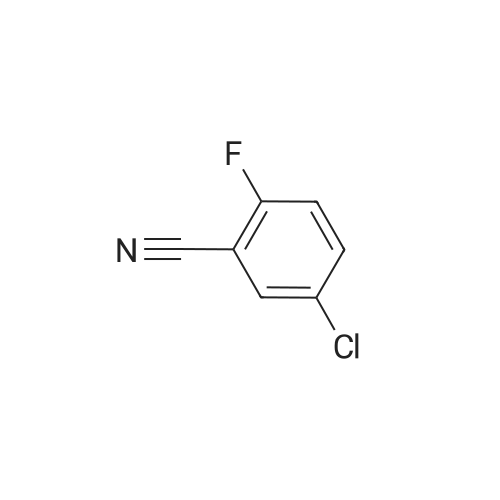
Chemistry
Chemistry- Boronic acids/esters >>1,3,2-DioxaborinaneAliphatic BoronsAromatic BoronsBCATBoric AcidsMIDA BoronatesOther BoronatesOther BoronsOxaborolesCatalysis Chemistry >>Achiral Crown LigandsC-H ActivationCarbon-Donor LigandsChiral Carbon LigandsChiral Crown LigandsChiral Olefin LigandsCross-CouplingCross-Coupling Using Transition Metal CatalystsDACH or Trost LigandsChemical Biology >>Conjugation Chemistry
- GlycoscienceLinkers and CrosslinkersNucleic Acid ChemistryPEGylationPeptide ChemistryPhotolabile Protecting GroupHeterocyclic Building Blocks >>AcridinesAliphatic HeterocyclesAromatic HeterocyclesAzetidinesBenzimidazolesBenzisoxazolesBenzodioxansBenzofuransBenzothiazolesInorganic reagents >>Inorganic ReagentOrganic Building Blocks >>Acid AnhydridesAcyl ChloridesAlcohols
- AldehydesAliphatic Chain HydrocarbonsAliphatic Cyclic HydrocarbonsAlkenesAlkylsAlkynesOrganometallic Reagents >>Grignard ReagentsOrganoarsenicOrganobismuthOrganoboronOrganogermaniumOrganolithiumOrganomercuryOrganosiliconOrganotinSpecialty Synthesis >>Enzyme-Mediated SynthesisFluorous SynthesisIonic LiquidsSolid Supported SynthesisStains and Dyes >>
- Acid DyesAzoic DyesBasic DyesDirect DyesDisperse DyesDye IntermediatesMordant DyesOil DyesOther Stains and DyesSynthetic Reagents >>Acids and BasesC-C Bond FormationC-X Bond Formation (Halogen)C-X Bond Formation (Non-Halogen)Chelation and Complexation CompoundsCondensation AgentsCouplingDehydrating ReagentsFluorination Reagent

 Pharmaceutical Intermediates
Pharmaceutical Intermediates- Analgesics >>BenzhydrocodoneBicifadineCapsaicinCelecoxibDebio-0827DexamethasoneElagolix SodiumEsketamine HydrochlorideIbuprofen SodiumAnesthetics >>CiprofolEsketamine HydrochlorideFospropofol Fospropofol DisodiumLevobupivacaine RemimazolamRemimazolam BesilateRemimazolam BesylateRopivacaineAnti-Addiction Agents >>Kakonein
- Lofexidine HydrochlorideVarenicline Anti-inflammatory Agents >>ApremilastAsunaprevirATB-346 RelatedBalsalazide BaricitinibBencycloquidium BromideBudesonideCefiderocol Sulfate TosylateCeftaroline Fosamil AcetateAntibacterials >>AFN-1252 RelatedAlatrofloxacin Bedaquiline FumarateBesifloxacin BrilacidinCaspofungin AcetateCefiderocol Sulfate TosylateCefilavancinCeftaroline Fosamil
- Anticonvulsants >>BrivaracetamCannabidiolEscitalopram OxalateEslicarbazepine Acetate RelatedFosphenytoinGabapentin EnacarbilJNJ-10234094LacosamideLevetiracetam RelatedAntidementia Agents >>Azeliragon RelatedDavunetideDonepezil HydrochlorideDonepezil RelatedEnsaculinFlorbetaben Flortaucipir F 18Flutemetamol IdalopirdineAntidepressants >>4-Chlorokynurenine
- AgomelatineAgomelatine RelatedAllopregnanolone RelatedAmibegronAmitifadineAripiprazole RelatedBasimglurantBefloxatoneAntiemetics >>AprepitantAprepitant RelatedDolasetron RelatedDolasetron Mesylate HydrateFosaprepitant DimeglumineFosaprepitant RelatedOlanzapinePalonosetron RelatedPalonosetron HydrochlorideAntifungals >>Anidulafungin RelatedCabotegravirMore >>

 Inhibitors/Agonists
Inhibitors/Agonists- ADC >>ADC AntibodyADC LinkerADC Linker with PayloadADC ToxinAntibody-Drug Conjugates (ADCs)Drug-Linker Conjugates for ADCAngiogenesis >>ALKBcr-AblBTKEGFRFAKFGFRFLT3HER2HIFAnti-infection >>3CLproAntibacterialAntibioticAntifungal
- AntiparasiticAntiprotozoalAntiviralArenavirusBVDVApoptosis >>Apoptosis InducerBcl-2Bcl-6BRKc-Mycc-RETC/EBPCARDCaspaseAutophagy >>Atg4ATG4BATTECsAUTACsAutophagyBeclin1
- FKBPLC3LRRK2Biochemical Reagent >>Cell Cycle >>AntifolateAPCAurora KinaseBUBCasein KinaseCdc25CDKChkCRISPR/Cas9Cytoskeleton >>ActinArp2/3 ComplexCYTHCytoplasmic DyneinDynaminFAKMore >>

 Material Science
Material Science- Aggregation-Induced Emission >>Other AIE BlocksTetraphenylethylene SeriesCOFs Linkers >>Aldehyde COFs LinkersAlkynyl COFs LinkersAmine COFs LinkersBoric COFs LinkersCyano COFs LinkersMultifunctional COFs linkersOther COFs linkersTriptycene SeriesElectronic Materials >>Battery MaterialsDye-Sensitized Solar Cell (DSSC) MaterialsElectronic MaterialLiquid Crystal (LC) MaterialsMolecular ConductorsOrganic Light-Emitting Diode (OLED) MaterialsOrganic Solar Cell (OPV) MaterialsOrganic Transistor (OFET) MaterialsPerovskite Solar Cell (PSC) MaterialsMagnetic Materials >>Magnetic Ionic LiquidsMagnetic MaterialMagnetic Metal Complexes
- Organic Radicals Material Chemical Compounds >>Ligands for Functional Metal ComplexesLiquid Crystal (LC) Building BlocksPhthalocyanine Building BlocksPolymer and Macromolecule Semiconductor Building BlocksSmall Molecule Semiconductor Building BlocksSolubility Enhancing Reagents Supramolecular Host Materials MOF Ligands >>Carboxylic Acid MOF LigandsCarboxylic Acid Nitrogen-Containing Mixed MOF LigandsNitrogen-Containing MOF LigandsOther MOF LigandsNanomaterials >>Carbon NanomaterialsCeramic MembranesDendrimersGold NanoparticlesIron Oxide NanoparticlesMaterials by ApplicationMesoporous MaterialsMetals and Metal AlloysNano Minerals: NanoclaysOLED Materials >>Dopant
- HostHTMOLED IntermediatesOther OLED related materialsOptical Materials >>Coumarin DyesCyanine and Squarylium DyesDCM DyesDipyrromethene DyesFluorescence MaterialsFluorescent ProbesHeat and Pressure Sensitive DyesNear-Infrared (NIR) DyesOrganic Non-Linear Optical (NLO) MaterialsOrganic Pigments >>Organic PigmentOther Materials >>Material OtherPolymer Science >>MonomersPolymer AdditivesPolymerization ReagentsPolymersResins








 For Research Only
For Research Only
 120K+ Compounds
120K+ Compounds
 Competitive Price
Competitive Price
 1-2 Day Shipping
1-2 Day Shipping