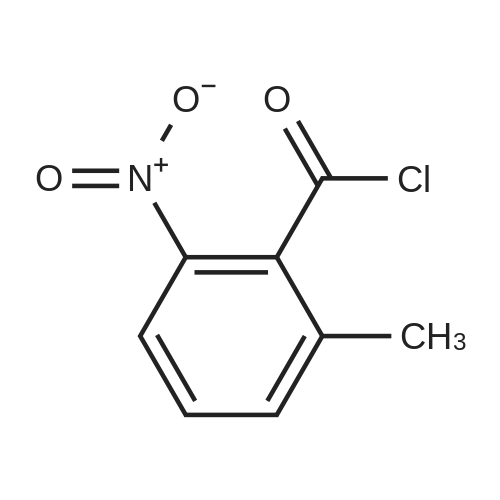
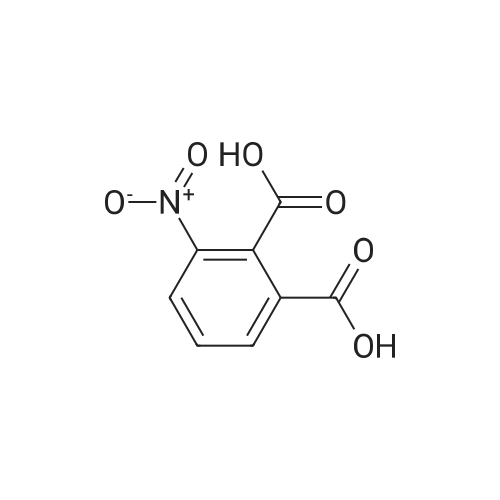
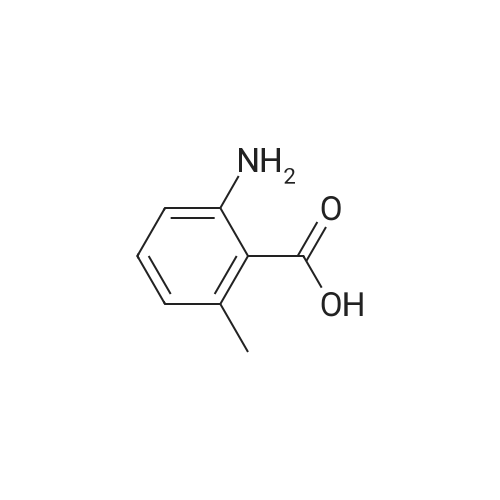
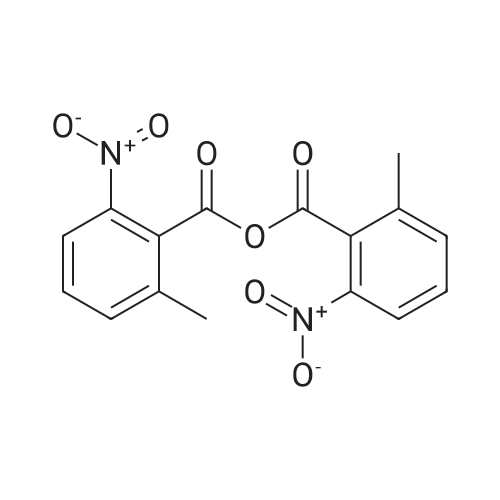
|
With potassium carbonate; In acetone; for 15h;Heating / reflux; |
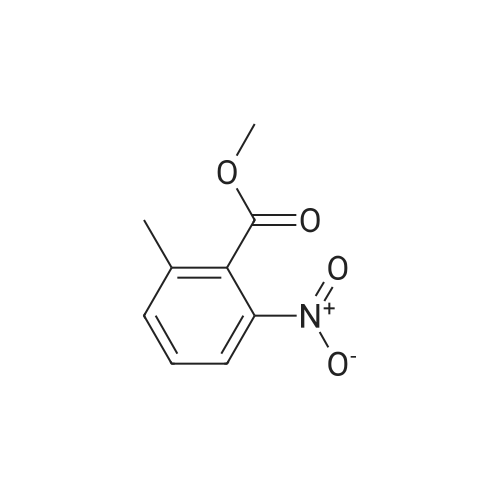
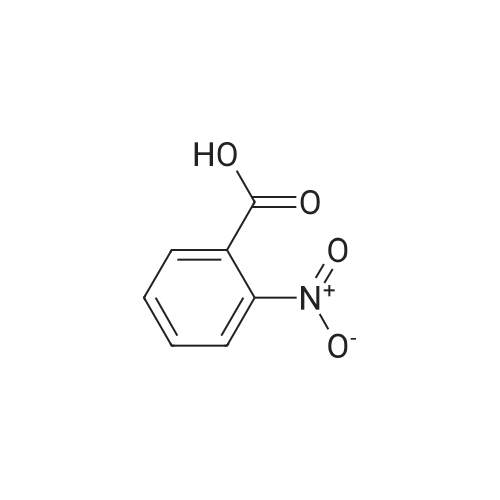
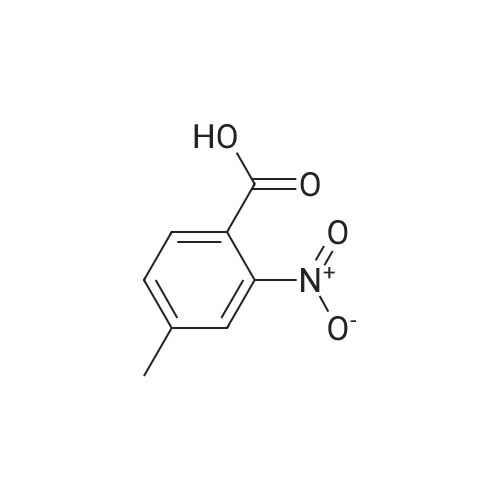
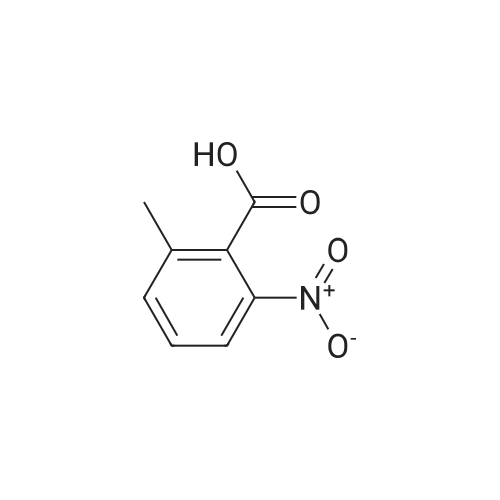
To a solution of the 2-methyl-6-nitro-benzoic acid (5.6 g, 30.9 mmol, 1.0 eq, Aldrich) and acetone (100 mL) was added crushed K2CO3 (21.4 g, 154.6 mmol, 5 eq) and MeI (9.6 mL, 154.6 mmol, 5.0 eq.). The reaction was heated to reflux for 15 h then cooled to RT, filtered and concentrated in vacuo. The filtrate was dissolved in EtOAc, washed with H2O and brine, dried (MgSO4) and concentrated in vacuo to give the desired compound as a reddish-brown oil which upon standing at RT crystallized as tan needles. |
|
With potassium carbonate; In acetone; for 15h;Heating / reflux; |
To a solution of 2-methyl-6-nitro-benzoic acid (5.6 g, 30.9 mmol, 1.0 eq, Aldrich) and acetone (100 mL) was added crushed K2CO3 (21.4 g, 154.6 mmol, 5 eq) and MeI (9.6 mL, 154.6 mmol, 5.0 eq). The reaction was heated to reflux for 15 h then cooled to RT, filtered and concentrated in vacuo. The filtrate was dissolved in EtOAc, washed with H2O and brine, dried (MgSO4) and concentrated in vacuo to give the desired compound as a reddish-brown oil which upon standing at RT crystallized to tan needles |
|
With potassium carbonate; In butanone; for 18h;Reflux; |
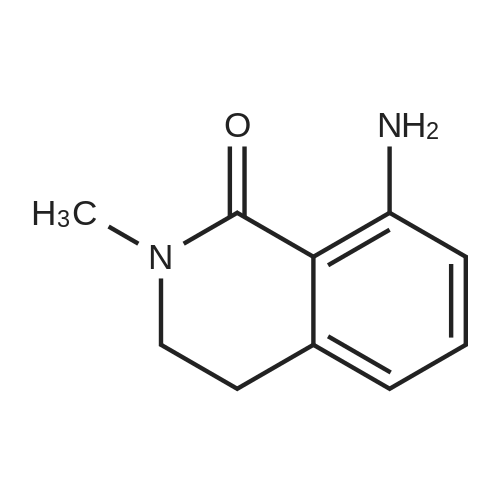
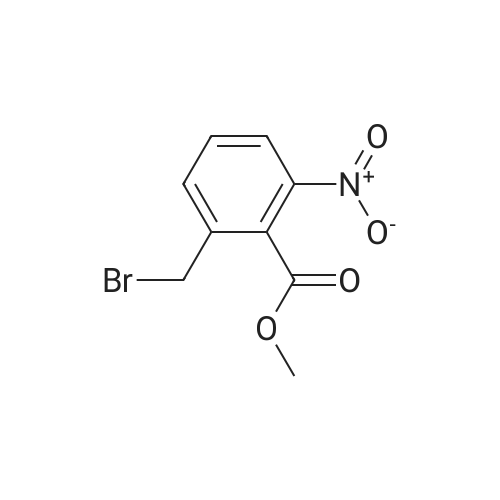
Example 1. Preparation of N-(3-oxo-2-(3-(trifluoromethyl)phenyl)isoindolin-4- yl)pyrazine-2-carboxamide (Compound 100):Step 1) Synthesis of methyl 2-methyl-6-nitrobenzoate (2):2-Methyl-6-nitrobenzoic acid (1; 10 g, 0.0552 mol) was taken up in 200 mL of methyl ethyl ketone along with methyl iodide (17.2 mL, 0.276 mol) and anhydrous potassium carbonate (38.1 g, 0.276 mol). The reaction mixture was stirred under reflux for 18 h. It was then cooled to room temperature and filtered. The filtrate was diluted with EtOAc (300 mL), washed with water (2 x 50 mL), brine, dried (Na2SO4) and concentrated under reduced pressure to afford methyl 2- methyl-6-nitrobenzoate 2 (10.80 g). |
|
With potassium carbonate; In butanone; for 18h;Reflux; |
Example 1. Preparation of N-(l-oxo-2-(3-(trifluoromethyl)phenyl)-l,2,3,4- tetrahydroisoquinolin-8-yl)pyrazine-2-carboxamide (Compound 101): Step 1) Synthesis of methyl 2-methyl-6-nitrobenzoate (2): 2-Methyl-6-nitrobenzoic acid (1; 10 g, 55.2 mol) was taken up in 200 mL of methyl ethyl ketone along with methyl iodide (17.2 mL, 276.0 mmol) and anhydrous potassium carbonate (38.1 g, 276.0 mmol). The reaction mixture was stirred under reflux for 18 h. It was then cooled to room temperature and filtered. The filtrate was diluted with EtOAc (300 mL), washed with water (2 x 50 mL), brine, dried (Na2SO4) and concentrated under reduced pressure to afford 10.80 g of methyl 2- methyl-6-nitrobenzoate 2. MS (ESI) calcd for C9H9NO4: 195.05; found: 196 [M + H]. |
| 10.80 g |
With potassium carbonate; In butanone; for 18h;Reflux; |
Step 1) Synthesis of methyl 2-methyl-6-nitrobenzoate (2) 2-Methyl-6-nitrobenzoic acid (1; 10 g, 55.2 mol) was taken up in 200 mL of methyl ethyl ketone along with methyl iodide (17.2 mL, 276.0 mmol) and anhydrous potassium carbonate (38.1 g, 276.0 mmol). The reaction mixture was stirred under reflux for 18 h. It was then cooled to room temperature and filtered. The filtrate was diluted with EtOAc (300 mL), washed with water (2*50 mL), brine, dried (Na2SO4) and concentrated under reduced pressure to afford 10.80 g of methyl 2-methyl-6-nitrobenzoate 2. MS (ESI) calcd for C9H9NO4: 195.05. found: 196 [M+H]. |






 Chemistry
Chemistry
 Pharmaceutical Intermediates
Pharmaceutical Intermediates
 Inhibitors/Agonists
Inhibitors/Agonists
 Material Science
Material Science







 For Research Only
For Research Only
 120K+ Compounds
120K+ Compounds
 Competitive Price
Competitive Price
 1-2 Day Shipping
1-2 Day Shipping