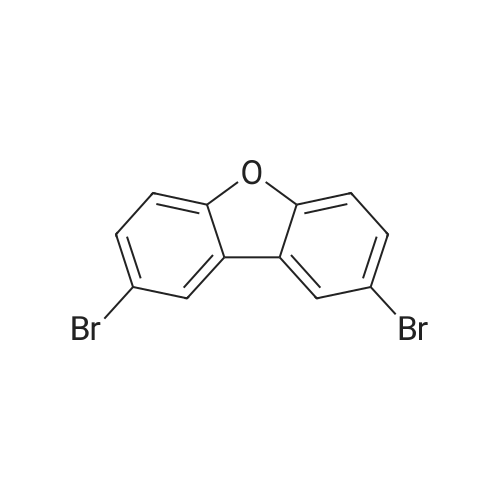
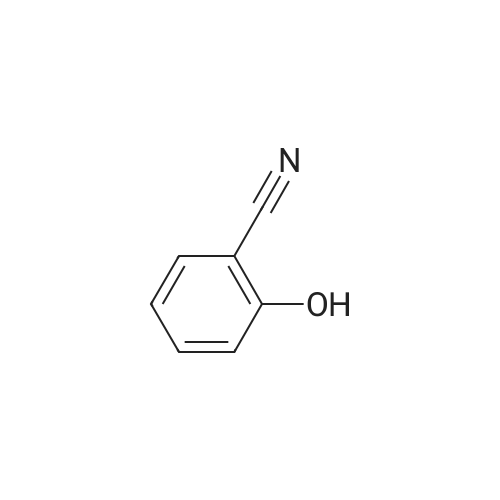
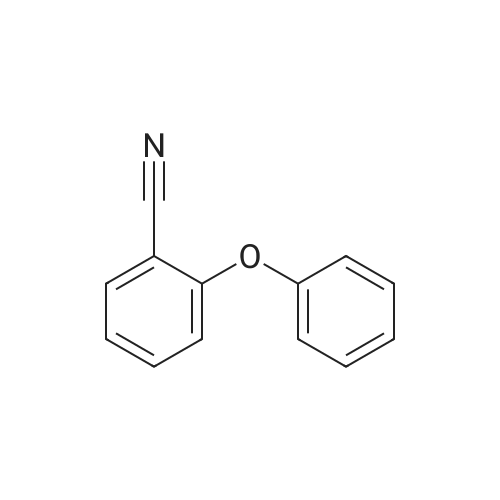
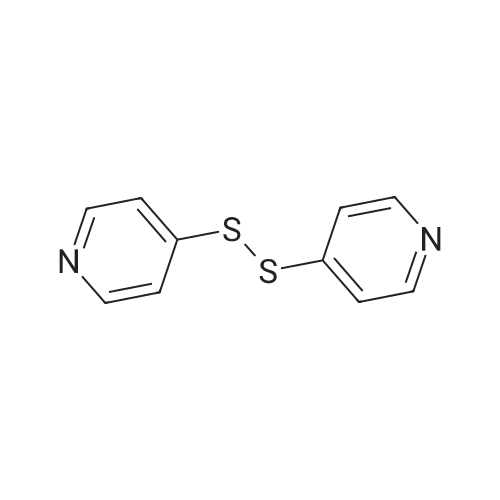
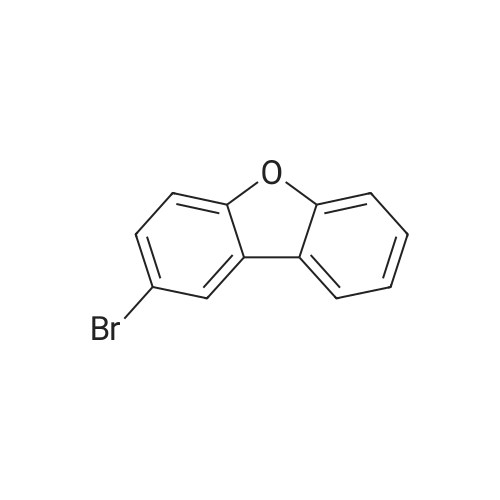
| 75.07% |
With N-Bromosuccinimide; In N,N-dimethyl-formamide; at 25℃; for 5h; |
(10 g, 59 mmol, 1.0 eq) was dissolved in 30 ml of dimethylformamide and slowly added at 25 C to a flask in which 30 ml of dibenzo [b, d] furan was dissolved in 30 ml of dimethylformamide, followed by addition of n-bromosuccinimide (21.69 g, 121 mmol, 2.05 eq) do.After 5 hours of reaction, the reaction mixture was precipitated with 200 ml of distilled water, filtered and dried to obtain 14.55 g of the title compound (yield: 75.07%). |
| 65% |
With bromine; In dichloromethane; at 0 - 20℃; for 12.7h;Inert atmosphere; |
(1) Synthesis of Compound (291-a) [0252] [0253] Into a three-necked flask, 168.1 g (1000 mmol) of dibenzofuran and 1600 ml of dichloromethane were charged, and the reaction vessel was cooled to 0 C in a nitrogen atmosphere. After adding 125 ml of a dichloromethane solution of 255.8 g of bromine dropwise into the reaction vessel over 40 min, the contents were stirred at room temperature for 12 h. After the reaction, the reaction vessel was cooled to 0 C and then 500 ml of water and 100 ml of a 20% aqueous solution of NaHSO4 were added. The resultant solution was extracted with several portions of dichloromethane in a separatory funnel. The extract was washed with 300 ml of a I N aqueous solution of sodium hydroxide, dried over anhydrous magnesium sulfate, filtered, and concentrated. The concentrate was dispersed in hexane for washing, to obtain a white solid. [0254] The yield was 212 g and the percent, yield was 65%. |
| 65% |
With bromine; In dichloromethane; at 0 - 20℃; for 12h;Inert atmosphere; |
(1) Synthesis of compound (140-a) [0243] [0244] In a three-neck flask, 168.1g (1000 mmol) of dibenzofuran and 1600 ml of dichloromethane were placed. The reactor was cooled to 0C in a nitrogen atmosphere. To the reactor, 125 mL of a dichloromethane solution of 255.8g of bromine was added dropwise over 40 minutes, and the resultant was stirred at room temperature for 12 hours. [0245] After completion of the reaction, the reactor was cooled to 0C. 500 mL of water was added, and further, 100 ml of a 20% aqueous NaHSO4 solution was added. The sample solution was transferred to a separating funnel, and extracted with dichloromethane several times. The resultant was washed with 300 ml of a 1 N aqueous sodium hydroxide solution and dried with anhydrous magnesium sulfate, filtrated and concentrated. The resulting product was washed by dispersing in hexane, whereby white solids were obtained. The yield was 212g (65%). |
| 40% |
With bromine; In tetrachloromethane; chloroform; at 20℃; for 168h; |
In a 500 mL three-neck flask were put 8.4 g (50 mmol) of dibenzofuran and 100 mL of carbon tetrachloride. A solution prepared by dissolving 17 g (110 mmol) of bromine in 50 mL of chloroform was dripped through a dropping funnel into the three-neck flask over about 20 minutes. Then, this solution was stirred at room temperature for 7 days. After that, this solution was washed with a saturated solution of sodium hydrogen carbonate, an aqueous solution of sodium thiosulfate and saturated brine. The organic layer was dried with magnesium sulfate, and this mixture was gravity filtered. The resulting filtrate was concentrated, and the obtained solid was recrystallized from chloroform. Accordingly, 6.4 g of a white powder was obtained in 40 % yield, which was the substance to be produced. |
| 38% |
With bromine; In acetic acid; at 75℃; for 3h; |
1a) 2,8-Dibromodibenzofuran Bromine (92.6 g, 0.58 mol) in acetic acid (54 g) is added at 75 C. to a solution of dibenzofuran (23.2 g, 0.14 mol) in acetic acid (232 g). The mixture is then stirred at 75 C. for 3 hours. The reaction mixture is cooled to room temperature and poured into H2O. The orange solid is washed with Na2S2O3 aq. and H2O. The crude product is then purified by recrystallization from n-hexane, wherein the pure product is obtained as a white solid (38% yield; mp.: 226 C.). 1H-NMR (CDCl3, ppm): 7.65 (d, 2H), 7.59 (dd, 2H), 8.03 (d, 2H). |
| 32% |
With bromine; acetic acid; In water; toluene; |
(1) Synthesis of 2,8-dibromodibenzofuran A three-necked flask was charged with dibenzofuran (100.91 g, 600 mmol) and 300 ml of AcOH and the contents were heated to 40 C. Then, a solution of Br2 (191.8 g, 1200 mmol)/AcOH 300 ml was added dropwise. After stirring for 9 h at 40 C., the mixture was refluxed for 6 h. After the reaction, the reaction production solution was cooled to room temperature and added with 600 ml of water. The precipitate collected by filtration was dissolved in toluene. The resultant solution was dried over anhydrous magnesium sulfate, filtrated and concentrated. The obtained solid product was recrystallized from hexane five times to obtain the titled compound (62.83 g, 32% yield). |
|
With bromine; In acetic acid; at 20℃;Reflux; Inert atmosphere; |
A 250 ml round bottom flask containing 8.4 g (50 mmol) of dibenzofuran dissolved in 100 ml of glacial acetic acid wasequipped with an addition funnel. Bromine 5.13 ml (100 mmol) in 30 ml of glacial acetic acid was added dropwise via the addition funnel to the dibenzofuran under constant stirring. This reaction mixture was stirred at room temperature for 4 h. It was then refluxed for 6 h, cooled. The solid was then collected by filtration and washed with three 100 ml portions of water. Recrystallization from 100 ml of acetic anhydride obtained 12.2 g (75%) pure 2,8-dibromodibenzofuran (1) as a white solid |
|
With bromine; In chloroform; at 25℃; for 48h; |
[0001082] To a solution of Compound 302A (1 g, 0.59 mmol) in CHC13 (6 mL) was added bromine (0.68 mL, 13.26 mmol) at 0 C. The mixture was stirred at 25 C for 48 h. It was poured into aq. Na2S203 (50 mL) and extracted with ethyl acetate (20 mL x 3). The combined organic layers were washed with brine (30 mL), dried over anhydrous sodium sulfate, and concentrated in vacuo to furnish the crude product. It was recrystallized from methanol to give Compound 302B. |






 Chemistry
Chemistry
 Pharmaceutical Intermediates
Pharmaceutical Intermediates
 Inhibitors/Agonists
Inhibitors/Agonists
 Material Science
Material Science







 For Research Only
For Research Only
 120K+ Compounds
120K+ Compounds
 Competitive Price
Competitive Price
 1-2 Day Shipping
1-2 Day Shipping