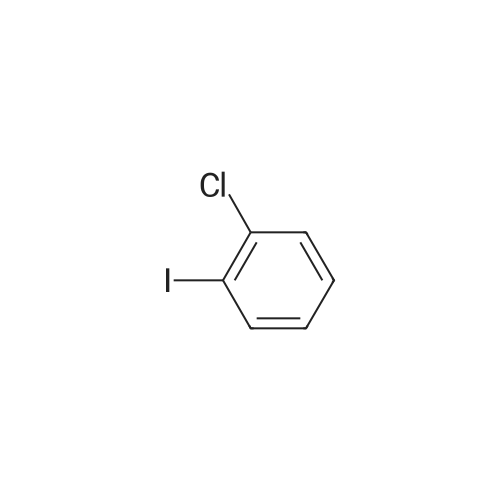
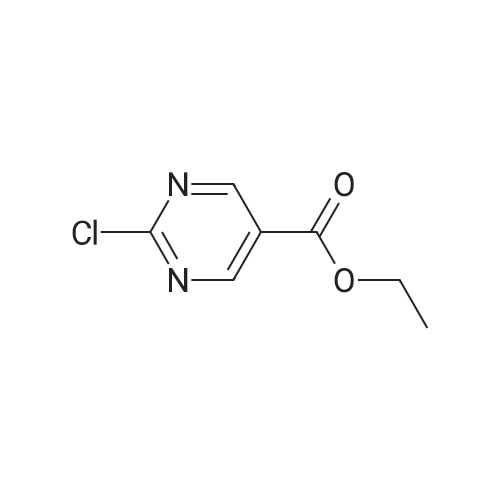
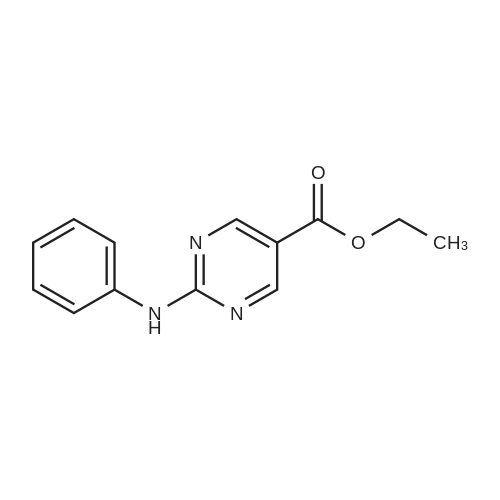
|
|
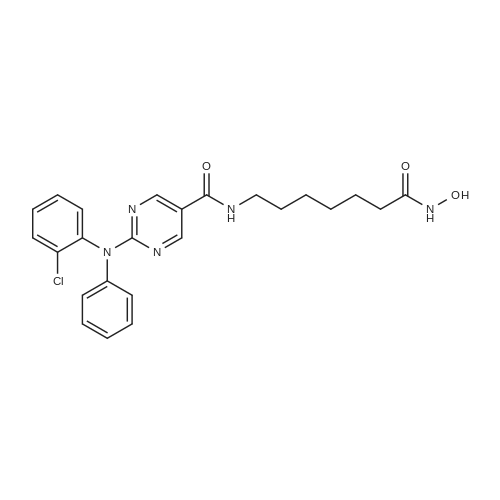
General procedure: Synthesis of 2-(diphenylamino)-N-(7-(hydroxyamino)-7-oxoheptyl)pyrimidine-5-carboxamide [0160] sodium hydroxide (2N, 20 mL) in MeOH (50 ml) and DCM (25 ml) was stirred at 0° C. for 10 min. Hydroxylamine (50percent) (10 ml) was cooled to 0° C. and added to the mixture. The resulting mixture was stirred at r.t. for 20 min. After removal of the solvent, the mixture was neutralized with 1M HCl to give a white precipitate. The crude product was filtered and purified by pre-HPLC to give a white solid (950 mg, 48percent). |
|
|
General procedure: A mixture of the compound 6 (2.0 g, 4.6 mmol), sodium hydroxide (2N, 20 mL) in MeOH (50 ml) and DCM (25 ml) was stirred at 0° C. for 10 min. Hydroxylamine (50percent) (10 ml) was cooled to 0° C. and added to the mixture. The resulting mixture was stirred at r.t. for 20 min. After removal of the solvent, the mixture was neutralized with 1M HCl to give a white precipitate. The crude product was filtered and purified by pre-HPLC to give a white solid (950 mg, 48percent). |
|
|
General procedure: A mixture of the compound 6 (2.0 g, 4.6 mmol), sodium hydroxide (2N, 20 mL) in MeOH (50 ml) and DCM (25 ml) was stirred at 0° C. for 10 min. Hydroxylamine (50percent) (10 ml) was cooled to 0° C. and added to the mixture. The resulting mixture was stirred at r.t. for 20 min. After removal of the solvent, the mixture was neutralized with 1M HCl to give a white precipitate. The crude product was filtered and purified by pre-HPLC to give a white solid (950 mg, 48percent). |
|
|
General procedure: Synthesis of 2-(diphenylamino)-N-(7-(hydroxyamino)-7-oxoheptyl)pyrimidine-5-carboxamide A mixture of the compound 6 (2.0 g, 4.6 mmol), sodium hydroxide (2N, 20 mL) in MeOH (50 ml) and DCM (25 ml) was stirred at 0° C. for 10 min. Hydroxylamine (50percent) (10 ml) was cooled to 0° C. and added to the mixture. The resulting mixture was stirred at rt for 20 min. After removal of the solvent, the mixture was neutralized with 1M HCl to give a white precipitate. The crude product was filtered and purified by pre-HPLC to give a white solid (950 mg, 48percent). |
|
With hydroxylamine; sodium hydroxide; In methanol; dichloromethane; at 0 - 20℃; for 0.333333h; |
General procedure: Synthesis of 2-(diphenylamino)-N-(7-(hydroxyamino)-7-oxoheptyl)pyrimidine-5 - carboxamide (Compound A): A mixture of the compound 6 (2.0 g, 4.6 mmol), sodiumhydroxide (2N, 20 mL) in MeOH (50 ml) and DCM (25 ml) was stirred at 0°C for 10 mm. Hydroxylamine (50percent) (10 ml) was cooled to 0°C and added to the mixture. The resulting mixture was stirred at r.t. for 20 mm. After removal of the solvent, the mixture wasneutralized with 1M HC1 to give a white precipitate. The crude product was filtered and purified by pre-HPLC to give a white solid (950 mg, 48percent). |
|
|
General procedure: A mixture of the compound 6 (2.0 g, 4.6 mmol), sodium hydroxide (2N, 20 mL) in MeOH (50 ml) and DCM (25 ml) was stirred at 0°C for 10mm. Hydroxylamine (50percent) (10 ml) was cooled to 0°C and added to the mixture. Theresulting mixture was stirred at rt for 20mm. After removal of the solvent, the mixture was neutralized with 1 M HC1 to give a white precipitate. The crude product was filtered and purified by pre-HPLC to give a white solid (950 mg, 48percent). |
|
|
General procedure: A mixture of the compound 6 (2.0 g, 4.6 mmol), sodium hydroxide (2N, 20 mL) in MeOH (50 ml) and DCM (25 ml) was stirred at 0 °C for lOmin. Hydroxylamine (50percent) (10 ml) was cooled to 0 °C and added to the mixture. The resulting mixture was stirred at rt for 20min. After removal of the solvent, the mixture was neutralized with 1M HC1 to give a white precipitate. The crude product was filtered and purified by pre-HPLC to give a white solid (950 mg, 48percent). |
|
|
General procedure: A mixture of the compound 6 (2.0 g, 4.6 mmol), sodium hydroxide (2N, 20 mL) in MeOH (50 ml) and DCM (25 ml) was stirred at 0 °C for 10 min. Hydroxylamine (50percent) (10 ml) was cooled to 0 °C and added to the mixture. The resulting mixture was stirred at r.t. for 20 min. After removal of the solvent, the mixture was neutralized with 1M HC1 to give a white precipitate. The crude product was filtered and purified by pre-HPLC to give a white solid (950 mg, 48percent). |
|
|
General procedure: Synthesis of 2-(diphenylamino)-N-(7-(hydroxyamino)- 7-oxoheptyl)pyrimidine-5- carboxamide: A mixture of the compound 6 (2.0 g, 4.6 mmol), sodium hydroxide (2N, 20 mL) in MeCH (50 ml) and DCM (25 ml) was stirred at 0°C for 10 mi Hydroxylamine (50percent) (10 ml) was cooled to 0°C and added to the mixture. The resulting mixture was stirred at r.t. for 20 mm. After removal of the solvent, the mixture was neutralized with 1M HClto give a whiteprecipitate. The crude product was filtered and purified by pre-HPLC to give a white solid (950 mg, 48percent). |
|
|
General procedure: A mixture of the compound 6 (2.0 g, 4.6 mmol), sodium hydroxide (2N, 20 ml_) in MeOH (50 ml) and DCM (25 ml) was stirred at 0 °C for 10 min. Hydroxylamine (50percent) (10 ml) was cooled to 0 °C and added to the mixture. The resulting mixture was stirred at r.t. for 20 min. After removal of the solvent, the mixture was neutralized with 1 M HCI to give a white precipitate. The crude product was filtered and purified by pre-HPLC to give a white solid (950 mg, 48percent). |
|
|
General procedure: A mixture of the compound 6 (2.0 g, 4.6 mmol), sodium hydroxide (2N, 20 ml_) in MeOH (50 ml) and DCM (25 ml) was stirred at 0 °C for 10 min. Hydroxylamine (50percent) (10 ml) was cooled to 0 °C and added to the mixture. The resulting mixture was stirred at r.t. for 20 min. After removal of the solvent, the mixture was neutralized with 1 M HCI to give a white precipitate. The crude product was filtered and purified by pre-HPLC to give a white solid (950 mg, 48percent) |






 Chemistry
Chemistry
 Pharmaceutical Intermediates
Pharmaceutical Intermediates
 Inhibitors/Agonists
Inhibitors/Agonists
 Material Science
Material Science







 For Research Only
For Research Only
 120K+ Compounds
120K+ Compounds
 Competitive Price
Competitive Price
 1-2 Day Shipping
1-2 Day Shipping